


在臭氧氧化处理染料废水的研究中,大多采用模拟废水,水质情况与实际生产过程中的废水差距较大。本研究选用染料生产工厂的实际生产废水,探究臭氧氧化的处理效果、氧化机理和降解路径,以期为染料生产企业的废水处理提供参考。
1 实验部分
1.1 废水的来源及性质
实验用水采用烟台某染料公司分散染料实际生产废水(经氧化钙混凝吸附工艺处理后的出水),水质指标:COD为4 291 mg/L,TOC为1 410 mg/L,总氮为511 mg/L,氨氮为23 mg/L,pH为8.5,色度为1 250倍。
1.2 实验方法
反应在自制的反应器(有效容积0.5 L,高径比10:1)中进行,首先加入废水50 mL,控制反应温度为25 ℃,通入臭氧连续曝气,臭氧发生器以纯氧作为气源,高压放电后得到臭氧和氧气的混合气体,臭氧通过反应器底部的微孔板以微气泡的形式均匀进入,稳定后定时取样,反应后的混合气体经过2%的KI和10%的NaOH溶液吸收后排出。采用NaOH和稀硫酸调节pH,通过控制反应时间调整臭氧投加量,探究pH和臭氧投加量对废水处理效果的影响。以废水紫外-可见吸收光谱特征指标(UV254、UV350、UV435)在氧化前后的去除率表征相应有机物结构的破坏效果;通过COD、TOC、总氮、氨氮、硝氮表征废水的处理效果。其中,COD采用微波消解重铬酸钾法测定;总氮、氨氮和硝氮采用国标法进行测定〔4〕;TOC采用TOC-VCPH型TOC仪(岛津)测定。
采用液相色谱-质谱分析处理过程中废水中有机物的变化。废水经0.22 μm滤膜过滤后,经自动进样器吸取10 μL样品,以0.2%甲酸和甲醇为流动相,打入液相色谱,经C18柱分离。通过质谱仪中的离子源攻击,使分子发生电离,进一步裂解为碎片离子,之后在电场和磁场的双重作用下,按照不同质荷比在电场和磁场中的偏转不同进行分离。最后用检测器检测,得到质谱图。
2 结果与讨论
2.1 pH对UV254、UV350、UV435去除率的影响
在臭氧投加量为136.8 g/L,pH分别为1、5.5、10的实验条件下,探究pH对臭氧氧化降解有机物效果的影响,以UV254、UV350、UV435去除率判断相应有机物结构的破坏效果,结果见表1。
表1 pH对UV254、UV350、UV435去除率的影响
| pH | UV254去除率/% | UV350去除率/% | UV435去除率/% |
| 1 | 46.50 | 84.25 | 98.81 |
| 5.5 | 49.60 | 88.18 | 98.83 |
| 10 | 57.00 | 88.00 | 99.03 |
根据紫外-可见吸收光谱分析结果,结合企业废水的实际情况,如果臭氧氧化进水pH过高,前段氧化钙混凝吸附时产生的硫酸钙渣较多,经济性较差,因此选取在pH=8.5的条件下,探究臭氧氧化处理分散染料废水的效率与机理。
2.2 臭氧对废水中有机物结构破坏效率
在臭氧投加量分别为0、1、2、3、4.5、9、13.5、18、36、72 g/L下进行实验,考察臭氧投加量对UV254、UV350、UV435的残留率和废水pH的影响,结果见图1。
图1
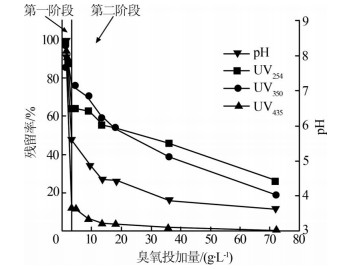
在碱性条件下,随着臭氧投加量的增加,废水颜色明显变淡,由深棕色变为浅黄色。由图1可知,当臭氧投加量为72 g/L时,UV254、UV350、UV435的残留率分别为26%、19%、1%,因此随着臭氧投加量的增加,UV254、UV350、UV435明显下降。说明在臭氧氧化过程中,废水中的染料生产副产物和中间产物中的偶氮双键断裂,助色基团和生色基团脱落。
在臭氧投加量达到3 g/L之前,特征指标下降得较快,之后下降速率较慢。因此可以以3 g/L为界,简单地将氧化过程划分为两个阶段。分别计算两个阶段的臭氧消耗量占总臭氧消耗量的比例(ω臭氧)、OH-变化量占总OH-变化量的比例(ω)和特征指标变
化量占总特征指标变化量的比例(ωμ),计算UV254、UV350、UV435在两个阶段的去除反应速率常数(K)和线性相关系数(R2),结果见表2。
表2 臭氧氧化两阶段废水特征指标变化统计数据
| 吸光度 | ωμ/% | ω臭氧/% | ωOH-/% | K/(L·g-1) | R2 | |
| 第一阶段 | UV254 | 48.37 | 4.17 | 99.85 | -0.002 2 | 0.925 8 |
| UV350 | 28.61 | -0.001 5 | 0.880 1 | |||
| UV435 | 88.26 | -0.005 4 | 0.730 6 | |||
| 第二阶段 | UV254 | 51.63 | 95.83 | 0.15 | -0.000 1 | 0.987 1 |
| UV350 | 71.39 | -0.000 2 | 0.941 6 | |||
| UV435 | 11.74 | -0.000 03 | 0.577 9 |
由表2可知,在第一阶段,UV254、UV350、UV435的ωμ分别为48.37%、28.61%、88.26%,仅消耗4.17%的臭氧,但消耗了99.85%的OH-;第二阶段,UV254、UV350、UV435的ωμ分别为51.63%、71.39%、11.74%,消耗了95.83%的臭氧和0.15%的OH-。在反应的第一阶段,UV254、UV350、UV435的氧化速率分别为第二阶段的22、7.5、180倍。
2.3 臭氧对废水中有机物的去除效果
在臭氧投加量分别为0、2、3、4.5、9、18、36、72 g/L下进行实验,考察不同臭氧投加量下废水的COD和TOC变化,结果见图2。
图2
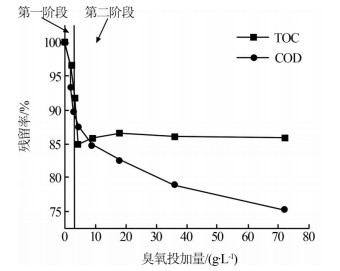
由图2可知,随着臭氧投加量的增加,废水的COD逐渐降低,TOC先下降到最低,然后又微弱地上升后保持稳定,这是因为差减法测定TOC时易受水中无机碳含量的影响,在反应前段,废水中有机物矿化产生的CO2在废水中积累,有机物矿化达到最大程度时,水中CO2浓度最大,导致TOC测定值偏低;随着体系pH的持续降低,TOC难以被去除,水中CO2逐渐被吹脱,使得TOC的测定值微弱上升,接近真实值〔9〕。根据上述紫外-可见吸收光谱的分析结果,废水的臭氧氧化过程可以分为两个阶段:在第一阶段,COD和TOC的去除率分别达到12.5%和15%;在第二阶段,当臭氧投加量达到72 g/L时,COD和TOC的去除率分别为25%和15%。TOC的去除在第一阶段就已经完成、COD第一阶段的去除速率为第二阶段的19倍。因此,臭氧氧化过程中废水中有机物的矿化发生在HO·作用的第一阶段,HO·氧化对有机物结构破坏和有机物的去除贡献更大,臭氧发挥氧化作用时难以实现有机物的矿化〔10〕。
2.4 含氮组分变化
在臭氧投加量分别为0、2、3、4.5、9、18、36 g/L的实验条件下,废水中含氮组分的变化见表3。
表3 废水中含氮组分的变化
| 臭氧投加量/(g·L-1) | 总氮/(mg·L-1) | 硝氮/(mg·L-1) | 氨氮/(mg·L-1) | 有机氮/(mg·L-1) |
| 0 | 511.12 | 0 | 22.53 | 488.60 |
| 1 | 489.43 | 0 | 43.86 | 445.58 |
| 2 | 522.91 | 0 | 43.16 | 479.76 |
| 4.5 | 510.90 | 54.18 | 42.41 | 414.32 |
| 9 | 503.17 | 70.34 | 33.77 | 399.06 |
| 18 | 514.24 | 106.14 | 15.28 | 392.81 |
| 36 | 548.33 | 138.20 | 10.44 | 399.70 |
| 72 | 564.24 | 125.56 | 1.00 | 437.68 |
由表3可知,在臭氧投加量为1 g/L时,氨氮达43.86 mg/L,之后随臭氧投加量的增加逐渐下降;在臭氧投加量为72 g/L时,降至1 mg/L。因此,在废水的臭氧氧化过程中,氨基易与有机物主体分离,进入水溶液中被氧化去除。原废水中硝氮量很低,随臭氧投加量的增加,硝氮量逐渐增加;臭氧投加量达到36 g/L时,硝氮达到138.20 mg/L;总氮含量略有上升,主要受检测方法影响;有机氮在臭氧投加量为18 g/L时降至392.81 mg/L,随后受总氮含量影响浓度轻微上升。
在染料废水的臭氧氧化过程中,染料中的氮元素主要是以氮气或硝态氮的形式被释放〔11〕。臭氧进入废水中生成的HO·发挥氧化作用,有机物中部分氨基受到HO·的攻击直接脱落进入水中,造成氨氮量短暂增加,进一步被氧化为硝酸根;染料中的偶氮键等以有机氮的形式存在于废水中,此类不饱和发色基团容易受到攻击而发生化学反应,被氧化为硝基或进一步生成硝酸根进入废水中,但仍有大量氮元素保留在有机物中。因此废水中氨氮和有机氮浓度下降,硝态氮浓度上升。总氮在测定过程中使用的消解液为碱性过硫酸钾,碱性过硫酸钾中的碱与废水中的氨氮会生成氨水,氨水挥发生成氨气和水,造成废水中总氮测定含量偏低。在氧化的前期,废水中氨氮含量较高,随着氧化过程的持续进行,废水中的氨氮被氧化为硝态氮,因此在氧化前期废水中总氮测定含量略低于氧化后期,表现出总氮含量略有上升,随之计算的有机氮含量也略有上升〔12〕。
2.5 降解路径分析
在臭氧投加量分别为0、3、9、72 g/L的实验条件下进行实验,采用液相色谱-质谱分析废水组分变化。通过谱图比对分析发现,氧化过程中废水的组分发生了明显的变化。在原废水中存在的物质种类较多,包含染料生产过程中的副产物及其结构类似物,含量较多的是3-羟基-5-硝基-2,1-苯并异噻唑、3-氨基-5-硝基-2,1-苯并异噻唑、N-乙基-N-氰乙氧乙基间甲苯胺。在臭氧氧化过程中,废水中的有机物组分明显减少,最先减少的是苯胺类物质,在臭氧投加量为3 g/L时,2种苯胺类染料生产副产物减少明显,苯并异噻唑类含量仍较高;在臭氧投加量达到9 g/L时,苯胺类物质几乎完全被氧化,苯并异噻唑类显著减少,同时,有3,5-二硝基-2,1-苯并异噻唑、对硝基苯酚和硝基甲苯等降解中间产物生成;在臭氧投加量达到72 g/L时,苯并异噻唑类几乎完全被氧化,废水中可以检测到仅有少量的苯胺类降解中间产物存在。
由废水的COD、TOC和紫外-可见吸收光谱检测结果可知,废水的吸光度下降明显,说明废水中有机物结构被破坏,但废水中的COD并未完全去除,且TOC去除比例明显少于COD去除比例,说明在臭氧氧化过程中,废水中染料中间体的生色基和助色基脱落,有机物结构改变,但有机物并未完全矿化。
3 结论
(1)紫外-可见吸收光谱分析结果表明,废水中芳香族有机物和偶氮键在碱性条件(pH=10)下更易被去除,苯并异噻唑类有机物在弱酸性条件(pH=5.5)下更易被去除,因此,可根据实际废水组分情况调整臭氧氧化初始pH,以获得最佳的处理效果。(2)碱性条件下的臭氧氧化过程分为两个阶段:第一阶段HO·发挥主要氧化作用,第二阶段臭氧发挥主要氧化作用。HO·对废水特征指标(UV254、UV350、UV435)的氧化速率分别约为臭氧的22、7.5、180倍。
COD和TOC的去除率为25%和15%,HO·对COD的去除速率为臭氧的19倍,而TOC的去除基本全部由HO·完成。因此在氧化过程中,废水中有机物结构破坏和矿化同时发生,HO·对有机物结构破坏和矿化的贡献程度更高。
(3)废水中氨氮和部分有机氮被氧化,转化为硝氮,废水中总氮并未达到明显的去除。
(4)废水中存在的主要有机物为苯胺类和苯并异噻唑类,在氧化过程中,有机物中氮组分被氧化为硝态氮,有机物的助色基和生色基脱落,结构被破坏,导致了废水中有机物结构破坏和矿化,氧化的中间产物主要为对硝基苯酚和硝基甲苯等,进一步生成有机酸、醇类,反而限制了HO·产率,使得氧化进程难以继续进行,限制了最终的氧化效率。
参考文献
Electrochemical degradation and toxicity reduction of C. I. Basic Red 29 solution and textile wastewater by using diamond anode
[J].
臭氧氧化与生物法联合处理苯酚和苯胺废水研究
[J].DOI:10.11894/1005-829x.2015.35(12).061 [本文引用: 1]
Degradation of acid orange 7 in aqueous solution by a novel electro/Fe2+/peroxydisulfate process
[J].DOI:10.1016/j.jhazmat.2012.02.047 [本文引用: 1]
Photochemistry of 5-nitro-1, 2-benzisothiazole derivatives:effects of substituents, solvents and excitation wavelength
[J].DOI:10.1016/j.tetlet.2008.03.104
A novel catalytic ceramic membrane fabricated with CuMn2O4 particles for emerging UV absorbers degradation from aqueous and membrane fouling elimination
[J].DOI:10.1016/j.jhazmat.2017.11.044 [本文引用: 1]
高级氧化技术处理染料废水的研究进展
[J].DOI:10.3969/j.issn.1005-829X.2006.06.001 [本文引用: 1]
Decolorization of textile basic dye in aqueous solution by ozone
[J].DOI:10.1016/j.dyepig.2011.07.012 [本文引用: 1]
Application of electrochemically generated ozone to the discoloration and degradation of solutions containing the dye reactive orange 122
[J].DOI:10.1016/j.jhazmat.2008.07.108 [本文引用: 1]
水体中总氮测定方法及相关问题的探讨
[J].DOI:10.3969/j.issn.1674-8409.2014.16.187 [本文引用: 1]
Mechanism and dynamic study of reactive red X-3B dye degradation by ultrasonic-assisted ozone oxidation process
[J].DOI:10.1016/j.ultsonch.2016.08.006 [本文引用: 1]


 津公网安备 12010602120337号
津公网安备 12010602120337号{kind=link}
{kind=link}
{kind=link}
{kind=link}