Metal-free catalytic ozonation on surface-engineered graphene: Microwave reduction and heteroatom doping
5
2019
... 随着工业化和城市化快速发展及世界人口增多,水资源需求量及消耗量快速增加,污染物排放至自然水体,致使水体使用功能降低,进一步加剧水资源供需不平衡;同时废水中含有大量难降解、有毒、致癌、致突变的有机污染物,严重威胁着人类健康与生态环境.因此需要采用有效的技术手段对其进行处理,使废水得到回收利用.用于废水处理的传统手段有生物法、物理法和化学法.但一些毒性大的有机污染物难以进行生物降解,一些难降解有机物在传统处理方法中降解效率又较低,难以取得理想的处理效果.高级氧化技术(AOPs)是基于羟基自由基(·OH)、硫酸根自由基(SO4·-)等高活性含氧物种(ROS)来氧化降解有机污染物的一种水处理技术,产生的·OH的氧化电势为+2.8 eV〔1〕,可高效去除较难氧化的有机物,克服传统水处理方法的限制. ...
... 臭氧氧化是一种主流的高级氧化技术,其利用臭氧做氧化剂对污染物进行氧化降解.臭氧(O3)的氧化电势为+2.07 eV〔1〕,具有较强的氧化能力,可在环境压力和温度下与多种有机物反应,在水处理中已经广泛用于去除气味和色度.臭氧氧化有机物的途径有2种,一是O3分子直接氧化水中有机污染物,二是O3分解产生ROS间接氧化有机污染物.但O3的氧化具有选择性,与部分有机物的反应较缓慢,通常只能攻击不饱和碳碳键,不能攻击饱和键,所以中间产物不能完全矿化,使得臭氧氧化有机物的矿化率较低;O3在水中分解产生ROS过程受溶液pH影响,随着pH升高而加快〔2〕,同时O3在水中较低的溶解度和稳定性降低O3的利用率.以上缺点导致臭氧氧化有机污染物的能力受到限制.研究发现使用催化剂可显著促进臭氧分解产生更多ROS,从而提高氧化降解有机污染物的能力.近年来,催化臭氧氧化技术已经成为处理工业废水领域的研究热点.催化臭氧氧化包括均相催化臭氧氧化和非均相催化臭氧氧化.由于均相催化臭氧氧化体系中催化剂难以回收,造成催化剂的损失与浪费,并且残留的催化剂会对水体产生二次污染,所以非均相催化臭氧氧化更具有适用性〔3〕.此外,非均相催化剂也可促进臭氧在气液两相之间的传质,提高臭氧利用率. ...
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... 〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... (2) 环加成反应.加成反应发生在不饱和键与亲电试剂之间,与之不同的是,O3与有机污染物之间的反应是通过环加成进行.R. Criegee〔31〕阐述O3与烯烃发生环加成反应的可能机理,如图 1〔1〕所示,该反应可分为三个步骤:臭氧与双键结构形成五元环;产生两性离子;在不同的反应途径中生成不同的产物,如酮、醛或酸. ...
The efficiency and mechanisms of catalytic ozonation
1
2010
... 臭氧氧化是一种主流的高级氧化技术,其利用臭氧做氧化剂对污染物进行氧化降解.臭氧(O3)的氧化电势为+2.07 eV〔1〕,具有较强的氧化能力,可在环境压力和温度下与多种有机物反应,在水处理中已经广泛用于去除气味和色度.臭氧氧化有机物的途径有2种,一是O3分子直接氧化水中有机污染物,二是O3分解产生ROS间接氧化有机污染物.但O3的氧化具有选择性,与部分有机物的反应较缓慢,通常只能攻击不饱和碳碳键,不能攻击饱和键,所以中间产物不能完全矿化,使得臭氧氧化有机物的矿化率较低;O3在水中分解产生ROS过程受溶液pH影响,随着pH升高而加快〔2〕,同时O3在水中较低的溶解度和稳定性降低O3的利用率.以上缺点导致臭氧氧化有机污染物的能力受到限制.研究发现使用催化剂可显著促进臭氧分解产生更多ROS,从而提高氧化降解有机污染物的能力.近年来,催化臭氧氧化技术已经成为处理工业废水领域的研究热点.催化臭氧氧化包括均相催化臭氧氧化和非均相催化臭氧氧化.由于均相催化臭氧氧化体系中催化剂难以回收,造成催化剂的损失与浪费,并且残留的催化剂会对水体产生二次污染,所以非均相催化臭氧氧化更具有适用性〔3〕.此外,非均相催化剂也可促进臭氧在气液两相之间的传质,提高臭氧利用率. ...
水溶液中均相催化臭氧氧化和多相催化臭氧氧化的比较
1
2015
... 臭氧氧化是一种主流的高级氧化技术,其利用臭氧做氧化剂对污染物进行氧化降解.臭氧(O3)的氧化电势为+2.07 eV〔1〕,具有较强的氧化能力,可在环境压力和温度下与多种有机物反应,在水处理中已经广泛用于去除气味和色度.臭氧氧化有机物的途径有2种,一是O3分子直接氧化水中有机污染物,二是O3分解产生ROS间接氧化有机污染物.但O3的氧化具有选择性,与部分有机物的反应较缓慢,通常只能攻击不饱和碳碳键,不能攻击饱和键,所以中间产物不能完全矿化,使得臭氧氧化有机物的矿化率较低;O3在水中分解产生ROS过程受溶液pH影响,随着pH升高而加快〔2〕,同时O3在水中较低的溶解度和稳定性降低O3的利用率.以上缺点导致臭氧氧化有机污染物的能力受到限制.研究发现使用催化剂可显著促进臭氧分解产生更多ROS,从而提高氧化降解有机污染物的能力.近年来,催化臭氧氧化技术已经成为处理工业废水领域的研究热点.催化臭氧氧化包括均相催化臭氧氧化和非均相催化臭氧氧化.由于均相催化臭氧氧化体系中催化剂难以回收,造成催化剂的损失与浪费,并且残留的催化剂会对水体产生二次污染,所以非均相催化臭氧氧化更具有适用性〔3〕.此外,非均相催化剂也可促进臭氧在气液两相之间的传质,提高臭氧利用率. ...
Activated carbon catalytic ozonation of oxamic and oxalic acids
1
2008
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Occurrence of both hydroxyl radical and surface oxidation pathways in N-doped layered nanocarbons for aqueous catalytic ozonation
2
2019
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... 〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Cerium, manganese and cobalt oxides as catalysts for the ozonation of selected organic compounds
1
2009
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
The role of cobalt oxide or magnesium oxide in ozonation of ammonia nitrogen in water
1
2020
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Cobalt modified red mud catalytic ozonation for the degradation of bezafibrate in water: Catalyst surface properties characterization and reaction mechanism
1
2016
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Mechanistic investigations of the pyridinic N-Co structures in Co embedded N-doped carbon nanotubes for catalytic ozonation
1
2021
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Catalytic ozonation of selected pharmaceuticals over mesoporous alumina-supported manganese oxide
1
2009
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Heterogeneous catalytic ozonation of ciprofloxacin in water with carbon nanotube supported manganese oxides as catalyst
1
2012
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Metal-free graphene-based catalyst-Insight into the catalytic activity: A short review
1
2015
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Carbon-based metal-free catalysts
1
2016
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Magnetic porous ferro-spinel NiFe2O4: A novel ozonation catalyst with strong catalytic property for degradation of di-n-butyl phthalate and convenient separation from water
4
2012
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... Yueming Ren等〔14〕通过溶胶-凝胶法制备NiFe2O4,研究发现O3与NiFe2O4的相互作用促进·OH的产生,邻苯二甲酸二丁酯(DBP)降解效率相比于单独臭氧氧化提升了2倍;NiFe2O4表面的羟基基团是臭氧与之相互作用的活性位点.Hui Zhao等〔20〕也通过实验证实表面羟基是NiFe2O4的活性位点,且比较NiFe2O4-H和NiFe2O4-C两种不同的处理过程得到的NiFe2O4的催化活性(NiFe2O4-H是NiFe2O4-P前驱体烘干制得,NiFe2O4-C是NiFe2O4-P前驱体800 ℃焙烧制得).研究发现,NiFe2O4-H具有更大的比表面积、孔体积及更高的表面羟基密度,可显著促进苯酚降解,而NiFe2O4-C却基本无法促进苯酚降解.研究还通过XPS对反应前后NiFe2O4进行表征,发现Ni2+/Ni3+与O2-/O2氧化还原循环在NiFe2O4与O3之间的电子传递过程中发挥重要的作用,并提出NiFe2O4催化O3产生·OH的可能途径,其机理如图 7所示. ...
... Ni2+/Ni3+与Co2+/Co3+是常见的氧化还原循环,Hai Chen等〔75〕首次合成NiCo2O4尖晶石并用于催化臭氧氧化体系.由于O3可选择性地攻击磺胺二甲嘧啶(SMT)中H-N键和苯环,所以臭氧可快速降解SMT,但无法完全矿化SMT,而在NiCo2O4尖晶石催化下可使TOC去除率提高3倍.该研究认为Ni和Co均在矿化过程中发挥作用,Co不直接参与氧化还原循环,而是作为Lewis酸性位点与吸附水分子作用产生表面羟基形成Ni(Ⅱ)Co2O4-O-H-O3电子传递桥梁,Ni2+用于提供电子使H-O和O-O键断裂产生HO2·和O2·-,与O3反应进一步产生·OH.除晶格氧之外〔14〕,该研究还发现产生的O2·-可还原Ni3+为Ni2+,保证多金属氧化物的持续催化能力. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Hierarchical shape-controlled mixed-valence calcium manganites for catalytic ozonation of aqueous phenolic compounds
2
2016
... 多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
... Other polymetallic oxides and their catalytic performance
Table 6 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Fe-Cu-O | 酸性红B(ARB) | ARB:90%(CO,60 min),66%(SO);COD:70%(CO,60 min),48%(SO) | O3 6 mg/min,100 mL/min;ARB 200 mg/L;催化剂1 g/L;pH 6.36 | 表面羟基 | 〔101〕 |
| Mn-Ce-O〔n(Mn)/n(Ce)=8/2〕 | 安替比林(AP) | AP:100%(CO,2 min),100%(SO,5 min);TOC:87%(CO,60 min),44%(SO,60 min) | O3 20 mg/L,200 mL/min;AP 40 mg/L;催化剂0.2 g/L;pH 6.5 | 表面电子传递 | 〔16〕 |
| Mn-Co-Fe-O | 对硝基苯酚(PNP) | PNP:100%(CO,15 min),100%(SO,60 min);TOC:95%(SO,60 min),50%(SO,60 min) | O3 2 mg/min;PNP 10 mg/L;催化剂0.1 g/L;pH 6.5 | 去质子化表面羟基(pH>pHpzc) | 〔103〕 |
| CaMn4O8 | 对硝基苯酚(PNP) | PNP:100%(CO,30 min),100%(SO,60 min) | O3 100 mg/min,50 mg/L;PNP 50 mg/L;催化剂0.1 g/L;pH 5.7 | Mn3+/Mn4+氧化还原循环促进O3分解 | 〔15〕 |
注: CO是催化臭氧氧化过程,SO是单独臭氧氧化过程. ...
Characterization and reactivity of Mn-Ce-O composites for catalytic ozonation of antipy-rine
2
2015
... 多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
... Other polymetallic oxides and their catalytic performance
Table 6 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Fe-Cu-O | 酸性红B(ARB) | ARB:90%(CO,60 min),66%(SO);COD:70%(CO,60 min),48%(SO) | O3 6 mg/min,100 mL/min;ARB 200 mg/L;催化剂1 g/L;pH 6.36 | 表面羟基 | 〔101〕 |
| Mn-Ce-O〔n(Mn)/n(Ce)=8/2〕 | 安替比林(AP) | AP:100%(CO,2 min),100%(SO,5 min);TOC:87%(CO,60 min),44%(SO,60 min) | O3 20 mg/L,200 mL/min;AP 40 mg/L;催化剂0.2 g/L;pH 6.5 | 表面电子传递 | 〔16〕 |
| Mn-Co-Fe-O | 对硝基苯酚(PNP) | PNP:100%(CO,15 min),100%(SO,60 min);TOC:95%(SO,60 min),50%(SO,60 min) | O3 2 mg/min;PNP 10 mg/L;催化剂0.1 g/L;pH 6.5 | 去质子化表面羟基(pH>pHpzc) | 〔103〕 |
| CaMn4O8 | 对硝基苯酚(PNP) | PNP:100%(CO,30 min),100%(SO,60 min) | O3 100 mg/min,50 mg/L;PNP 50 mg/L;催化剂0.1 g/L;pH 5.7 | Mn3+/Mn4+氧化还原循环促进O3分解 | 〔15〕 |
注: CO是催化臭氧氧化过程,SO是单独臭氧氧化过程. ...
Mechanistic evaluation of ferrite AFe2O4(A=Co, Ni, Cu, and Zn)catalytic performance in oxalic acid ozonation
6
2017
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
... 不同的A位金属也会影响尖晶石多金属氧化物在催化反应中的活性.Fengzhen Zhang等〔17〕通过溶胶-凝胶法合成CoFe2O4、CuFe2O4、NiFe2O4和ZnFe2O4,并比较它们催化臭氧氧化草酸的性能.结果表明,这四种尖晶石多金属氧化物均能促进草酸的降解,120 min内TOC去除率分别为68.3%、15%、15%、8.5%,高于无催化剂时的4.7%.CoFe2O4催化臭氧分解草酸有最高的反应速率常数,得益于Co2+-Co3+-Co3+氧化还原循环较强的电子传递能力.除此之外,研究还发现AFe2O4表面高价态A金属离子含量和表面羟基密度与臭氧降解反应速率常数成正相关,且通过X射线光电子能谱(XPS)分析发现Co与Fe之间存在着协同作用,进一步促进臭氧的分解;最后提出AFe2O4-O3-草酸基于自由基及非自由基的降解路径,见图 8. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
... 〔
17〕
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
... 〔
17〕
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
... 〔
17〕
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Catalytic ozonation for advanced treatment of incineration leachate using(MnO2-Co3O4)/ACs a catalyst
1
2017
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Enhanced sulfame-thoxazole ozonation by noble metal-free catalysis based on magnetic Fe3O4 nanoparticles: Catalytic performance and degradation mechanism
1
2016
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Novel magnetically separable nanomaterials for heterogeneous catalytic ozonation of phenol pollutant: NiFe2O4 and their performances
4
2013
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... 尖晶石铁氧体MFe2O4(M=Mn2+、Ni2+、Co2+、Cu2+、Zn2+)是催化臭氧氧化体系应用最广泛也是研究最多的尖晶石氧化物,具有高密度的表面羟基(Me-OH)〔69〕,大量的氧空位,有丰富且廉价的原料来源〔63〕.除此之外尖晶石铁氧体还具有磁性,在完成催化反应后,可通过磁吸工艺把磁性颗粒从溶液中分离出来〔69〕.Hui Zhao等〔20〕以水热法合成具有磁性的NiFe2O4尖晶石铁氧体,将其用于催化臭氧氧化苯酚,并利用其自带的顺磁性成功分离纳米颗粒与处理后的废水溶液. ...
... Yueming Ren等〔14〕通过溶胶-凝胶法制备NiFe2O4,研究发现O3与NiFe2O4的相互作用促进·OH的产生,邻苯二甲酸二丁酯(DBP)降解效率相比于单独臭氧氧化提升了2倍;NiFe2O4表面的羟基基团是臭氧与之相互作用的活性位点.Hui Zhao等〔20〕也通过实验证实表面羟基是NiFe2O4的活性位点,且比较NiFe2O4-H和NiFe2O4-C两种不同的处理过程得到的NiFe2O4的催化活性(NiFe2O4-H是NiFe2O4-P前驱体烘干制得,NiFe2O4-C是NiFe2O4-P前驱体800 ℃焙烧制得).研究发现,NiFe2O4-H具有更大的比表面积、孔体积及更高的表面羟基密度,可显著促进苯酚降解,而NiFe2O4-C却基本无法促进苯酚降解.研究还通过XPS对反应前后NiFe2O4进行表征,发现Ni2+/Ni3+与O2-/O2氧化还原循环在NiFe2O4与O3之间的电子传递过程中发挥重要的作用,并提出NiFe2O4催化O3产生·OH的可能途径,其机理如图 7所示. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Mechanism and kinetics of catalytic ozonation for elimination of organic compounds with spinel-type CuAl2O4 and its precursor
3
2019
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... Yin Xu等〔21〕通过溶胶-凝胶法合成CuAl2O4尖晶石并使用一种偶氮染料酸性橙7(AO7)来研究它的催化性能及可能的反应机理.结果表明CuAl2O4可提高AO7的降解率(96%,单独臭氧氧化76%)和COD去除率(87.2%,臭氧氧化40%);降解效果反映出CuAl2O4更多地是促进中间体的矿化.研究证实催化剂表面的·OH(吸附的·OH)是主要的ROS,除·OH之外,O2·-也参与AO7及其中间体的降解;在ROS产生过程中,≡Cu2+与≡Al3+有着协同作用,≡Al3+可作为臭氧和有机物吸附的活性位点(表面羟基和路易斯酸性位点)的储库,而≡Cu2+/≡Cu+氧化还原循环充当催化剂与O3之间电子传递的桥梁(图 11).该研究还发现电中性(pH≈pHpzc)的表面羟基最有利于羟基自由基的产生;除此之外,质子化(pH < pHpzc)的表面相比于去质子化(pH > pHpzc)的催化剂表面有更高的催化活性,这是因为臭氧的亲核性和亲电性使其更倾向于结合H而不是O. ...
... 多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Heterogeneous catalytic degradation of phenolic substrates: Catalysts activity
1
2009
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Fe-based catalysts for heterogeneous catalytic ozonation of emerging contaminants in water and wastewater
3
2017
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... (3) 链终止.·OH的氧化是非选择性的,可与大部分有机污染物和一部分无机物反应.臭氧在酸性和中性溶液中较稳定,碱性溶液中易分解,所以·OH形成也会受到pH限制;体系中过多的O3及自由基也会导致链反应的终止,见式(6)、式(7)〔23〕.除此之外溶液中存在的无机阴离子是自由基的猝灭剂,比如HCO3-,CO32-,HPO42-和H2PO4-等会快速与·OH等自由基反应,造成自由基的猝灭〔34〕. ...
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
Catalytic ozonation for water and waste-water treatment: Recent advances and perspective
4
2020
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
... O3直接氧化和O3分解产生ROS间接氧化是臭氧与有机污染物反应的两种主要作用机理〔24〕.O3和·OH与一些常见有机物的表观二级反应速率常数列于表 1〔26-29〕. ...
... (2) 链增长.在臭氧不充足的情况下,生成的HO2·和O2·-与O2和HO2-不成比例,此时会导致H2O2生成.在O3充足情况下通过进一步反应,体系会产生·OH,见式(3)~式(5)〔24〕. ...
... 非均相催化剂可提高臭氧的利用率,一方面可提高臭氧的传质效率,另一方面可促进臭氧分解产生更多的活性氧物种,从而提高臭氧的氧化能力;非均相催化臭氧氧化涉及多个反应和步骤,并且受多种因素影响.催化臭氧氧化体系包括气相(O3)、水相和固体(催化剂)三相,因此很难对其反应机理进行详细的研究和理解.催化剂在催化臭氧氧化过程中可发挥多种作用,如为臭氧的吸附和分解提供反应场所.因此,可根据催化剂与O3和有机物的相互作用,划分出如图 2所示的3种情况〔24〕: ...
Ce-based catalysts used in advanced oxidation processes for organic wastewater treatment: A review
1
2020
... 用于催化臭氧氧化体系的非均相催化剂主要有两大类,一是非金属材料,包括活性炭〔4〕、碳纳米管〔5〕、还原氧化石墨烯〔1〕等纳米碳材料,二是金属基材料,包括简单金属氧化物〔6-7〕、负载金属〔8-9〕和负载金属氧化物〔10-11〕材料及钙钛矿氧化物、尖晶石氧化物、天然矿石、羟基氧化物等多金属氧化物.碳基材料往往具有较高的比表面积〔12〕,在催化臭氧氧化中有较高的活性〔5〕,而且制备成本更低,来源广泛〔13〕,但在反应过程中容易失活;相比于碳基材料,金属基材料尤其是过渡金属氧化物中多变的价态及表面存在的路易斯酸性位点促进O3的吸附和电子传递〔14-18〕,在催化臭氧氧化中有更高的活性.此外,一些金属催化剂还具有磁性,易于反应后的回收〔19-20〕,但在臭氧和ROS存在的环境中,金属离子的溶出也可能会产生二次污染.在金属基材料中,相比于简单金属氧化物,钙钛矿、尖晶石等多金属氧化物中由于不同金属的氧化还原循环之间存在着协同作用,催化臭氧分解的活性往往高于单一组分金属氧化物〔21〕,并且稳定性有所提升.近年来,MnO2、Al2O3、TiO2及Fe、Ce、Co的氧化物被广泛应用到催化臭氧氧化体系,有学者综述了它们在催化臭氧中的应用〔22〕.Jianlong Wang等〔23-24〕综述了Fe-基催化剂及其他金属氧化物在催化臭氧中的应用;Yuxian Wang等〔1〕综述了催化臭氧中常用的催化剂,主要介绍金属基的催化剂在催化臭氧中的应用;Lijun Niu等〔25〕综述了Ce-基催化剂在高级氧化中的应用,重点介绍在催化臭氧氧化技术中的应用.这些综述重点总结了常见的简单金属氧化物及金属基负载催化剂在催化臭氧氧化技术中的应用及反应机理,缺少对不同类型多金属氧化物在催化臭氧氧化中的应用及活性位点、催化机理的总结. ...
Catalytic ozonation: A promising advanced oxidation technology for water treatment
1
1999
... O3直接氧化和O3分解产生ROS间接氧化是臭氧与有机污染物反应的两种主要作用机理〔24〕.O3和·OH与一些常见有机物的表观二级反应速率常数列于表 1〔26-29〕. ...
Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅱ: Dissociating organic compounds
0
1983
Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅰ: Non-dissociating organic compounds
0
1983
Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals(·OH/·O-)in aqueous solution
1
1988
... O3直接氧化和O3分解产生ROS间接氧化是臭氧与有机污染物反应的两种主要作用机理〔24〕.O3和·OH与一些常见有机物的表观二级反应速率常数列于表 1〔26-29〕. ...
1
... (1) 氧化还原反应.臭氧有着较高的氧化还原电位,是一种较强的氧化剂,可直接和一些有机污染物发生氧化还原反应,这些氧化还原反应主要是通过电子转移过程进行〔30〕. ...
Mechanism of ozonolysis
1
1975
... (2) 环加成反应.加成反应发生在不饱和键与亲电试剂之间,与之不同的是,O3与有机污染物之间的反应是通过环加成进行.R. Criegee〔31〕阐述O3与烯烃发生环加成反应的可能机理,如图 1〔1〕所示,该反应可分为三个步骤:臭氧与双键结构形成五元环;产生两性离子;在不同的反应途径中生成不同的产物,如酮、醛或酸. ...
Nanocarbon-based catalytic ozonation for aqueous oxidation: Engineering defects for active sites and tunable reaction pathways
1
2020
... 溶液中的溶解O3可分解产生ROS,比如·OH、O2·-或者单线态氧1O2.相比于O3,·OH的氧化没有选择性且具有更高的氧化电势〔32〕(见表 2),可快速地和多种有机污染物反应.链引发、链增长、链终止是臭氧产生自由基的公认途径. ...
The reaction of ozone with the hydroxide ion: Mechanistic considerations based on thermokinetic and quantum chemical calculations and the role of HO4· in superoxide dismutation
1
2010
... (1) 链引发.臭氧分解产生·OH等ROS的链引发过程涉及式(1)、式(2)〔33〕. ...
Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment
1
2003
... (3) 链终止.·OH的氧化是非选择性的,可与大部分有机污染物和一部分无机物反应.臭氧在酸性和中性溶液中较稳定,碱性溶液中易分解,所以·OH形成也会受到pH限制;体系中过多的O3及自由基也会导致链反应的终止,见式(6)、式(7)〔23〕.除此之外溶液中存在的无机阴离子是自由基的猝灭剂,比如HCO3-,CO32-,HPO42-和H2PO4-等会快速与·OH等自由基反应,造成自由基的猝灭〔34〕. ...
Perovskites as substitutes of noble metals for heterogeneous catalysis: Dream or reality
1
2014
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
Lanthanum-based perovskites as catalysts for the ozonation of selected organic compounds
4
2013
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
... 〔36, 41-42〕和共沉淀法〔37〕. ...
... 这些早期的研究证明钙钛矿在催化臭氧氧化体系的可用性和高效性,但对于活性氧物种及自由基的鉴定与分析缺乏相关材料支持,对钙钛矿的结构、臭氧与钙钛矿之间的相互作用机制更是缺少系统研究.因为A位金属在钙钛矿结构中起支撑作用〔36〕,所以本研究根据不同的A位金属,分别综述LnBO3、SrBO3(Ln为镧系金属元素,B为过渡金属元素,下同)在催化臭氧氧化中的应用及反应机理. ...
... 镧系元素的原子半径非常有利于ABO3立方晶格的形成〔49〕,且镧系金属阳离子可促进B位金属阳离子在催化反应中的暴露并且不影响催化活性,因此镧系元素(主要是La、Ce、Pr)作为钙钛矿的A位金属受到广泛的研究.C. A. Orge等〔36〕研究不同B位金属的镧基钙钛LaBO3(B=Fe、Ni、Co、Mn)在催化臭氧氧化中的活性.草酸是常见的有机物氧化中间体之一,臭氧很难将其降解,C. A. Orge等发现,LaBO3(B=Fe、Ni、Co、Mn)能促进臭氧对草酸的降解,其催化活性LaMnO3 > LaCoO3 > LaNiO3 > LaFeO3 > 仅臭氧;通过对LaMnO3/O3/草酸体系进一步研究发现,除溶液相中的·OH外,表面反应在催化臭氧降解草酸过程中也发挥重要的作用.更进一步,虽然LaCoO3的BET比表面积比LaMnO3低一倍,但两者几乎有同样的催化效果,这得益于LaCoO3表面存在大量的氧空位,从而证实氧空位对催化活性的重要作用. ...
Role of oxygen vacancies and Mn sites in hierarchical Mn2O3/LaMnO3-δ perovskite composites for aqueous organic pollutants decontamination
5
2019
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
... 〔37〕. ...
... 然而,并非在所有钙钛矿催化臭氧氧化有机物过程中,·OH都起主导作用,也可能存在其他ROS,或者还存在非自由基反应.Yuxian Wang等〔37〕研究LaMn4Ox(Mn2O3与LaMnO3-δ混合氧化物,Mn2O3质量分数14%)催化臭氧氧化苯并三氮唑(BTA)体系中的ROS.除·OH,体系中超氧自由基O2·-及非自由基ROS-1O2也是降解BTA的活性氧物种.其通过密度泛函理论(DFT)揭示O3在钙钛矿LaMnO3-δ表面的吸附和作用机理:(1)氧空位、Mn4+/Mn3+氧化还原中心及表面羟基都是O3吸附分解的潜在活性位点,与臭氧之间的吸附能分别为-2.04、-1.48、-0.46 eV,氧空位具有最大的吸附能,说明有更高的催化潜力.(2)氧空位可促进电子在催化剂与臭氧之间传递,O3会自发地在氧空位上分解产生双氧分子O2*.(3)表面Mn和表面羟基-OH可拉长O3中的O-O键从而促进O3分解.以上结论均采用实验表征进行验证,通过O2-TPD探究LaMn4Ox和LaMnOx表面的氧空位,发现提高Mn/La比有助于形成更多的氧空位,并且氧空位的存在有利于催化反应;更进一步,使用碘滴定法对氧空位含量进行测定,发现氧空位(δ值)越大,催化臭氧氧化BTA的反应速率常数越大.在此基础上,提出臭氧与LaMn4Ox的作用机理,如图 5所示. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
Oxidative nonstoichiometry in perovskites, an experimental survey; the defect structure of an oxidized lanthanum manganite by powder neutron diffraction
1
1974
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
Anion charge storage through oxygen intercalation in LaMnO3 perovskite pseudocapacitor electrodes
1
2014
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
La1-xCaxCoO3 perovskite-type oxides: Identification of the surface oxygen species by XPS
1
2006
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
Study of the formation of LaMO3(M=Co, Mn)perovskites by propionates precursors: Application to the catalytic destruction of chlorinated VOCs
1
2001
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
Synchronously degradation benzotriazole and elimination bromate by perovskite oxides catalytic ozonation: Performance and reaction mechanism
3
2018
... 钙钛矿多金属氧化物通式为ABO3,它具有丰富的组成和稳定且可调的晶体结构,可容纳元素周期表中90%以上的金属元素〔35〕.其结构中的A位金属起结构支撑作用,主要是碱土金属、碱金属或稀土金属的阳离子,一般为Sr、Ba、La、Ce、Pr,其位于钙钛矿立方体结构的中心,与12个O配位;B位金属是钙钛矿在催化反应中的活性位点,对于催化性能的调节至关重要〔36-37〕,B位主要是有电子在3d,4d和5d轨道分布的过渡金属离子,如Mn、Co、Ni、Fe,其位于钙钛矿结构立方体的顶角,与6个O配位〔38〕.图 3是钙钛矿的晶体结构〔39〕.与常见的多金属氧化物相比,钙钛矿氧化物对于在A位和B位进行元素部分取代有较强的适应性,且部分A位或者B位取代可改变钙钛矿的物理化学性质,如A位部分取代可改变B位金属的氧化态,同时会带来结构缺陷,B位部分取代可提高钙钛矿结构的稳定性及催化活性〔40〕.研究中常用的钙钛矿合成方法有溶胶-凝胶法〔36, 41-42〕和共沉淀法〔37〕. ...
... 通过相同或不同价态的其他阳离子C或D部分取代A位或B位元素可形成取代型的钙钛矿A1-xCxB1-yDyO3(0≤x≤1),可能会改变B位金属氧化态或造成结构缺陷,比如氧空位;除此之外,部分取代B位金属还可提高钙钛矿的稳定性和催化活性〔42〕.在一些非金属材料如石墨烯〔53〕、石墨相氮化碳(g-C3N4)〔54〕的研究中也报道结构缺陷有促进臭氧分解的作用.研究证实提高La1-xCaxCoO3钙钛矿中Ca掺杂量会导致表面氧空位出现〔42〕.少量Ni取代B位金属Fe生成的LaFe0.95Ni0.05O3比LaFeO3有更高的催化臭氧分解的能力〔55〕.C. A. Orge使用Cu部分取代B位金属Fe合成LaFe0.7Cu0.3O3和LaFe0.9Cu0.1O3钙钛矿,其催化臭氧氧化草酸的活性较未取代的LaFeO3明显降低;但通过Cu部分取代A位La元素合成的La0.8Ce0.2Al0.7Cu0.3在催化臭氧氧化草酸的研究中取得比LaAl0.7Cu0.3更好的效果.该工作总结得出,催化剂表面不仅需要臭氧的吸附位点,比如氧空位,B位金属对氧化能力的调节也很重要,所以还需要更深入的研究. ...
... 〔42〕.少量Ni取代B位金属Fe生成的LaFe0.95Ni0.05O3比LaFeO3有更高的催化臭氧分解的能力〔55〕.C. A. Orge使用Cu部分取代B位金属Fe合成LaFe0.7Cu0.3O3和LaFe0.9Cu0.1O3钙钛矿,其催化臭氧氧化草酸的活性较未取代的LaFeO3明显降低;但通过Cu部分取代A位La元素合成的La0.8Ce0.2Al0.7Cu0.3在催化臭氧氧化草酸的研究中取得比LaAl0.7Cu0.3更好的效果.该工作总结得出,催化剂表面不仅需要臭氧的吸附位点,比如氧空位,B位金属对氧化能力的调节也很重要,所以还需要更深入的研究. ...
Effects of different catalysts on the ozonation of pyruvic acid in water
2
2006
... F. Beltran课题组〔43〕通过溶胶-凝胶法合成不同钙钛矿氧化物LaTi1-xMexO3(Me=Cu,Co),包括LaTi0.15Cu0.85O3、LaTi0.4Cu0.6O3、LaTi0.78Cu0.22O3、LaTi0.15Co0.85O3、LaTi0.4Cu0.6O3,用于催化臭氧氧化丙酮酸.研究发现LaTi1-xMexO3均能提高O3的氧化能力,且LaTi0.15Cu0.85O3具有最好的催化性能.F. Beltran课题组进一步使用LaTi0.15Cu0.85O3钙钛矿催化臭氧氧化丙酮酸〔44〕和没食子酸〔45〕,系统地研究操作条件,包括臭氧浓度、催化剂用量、温度、pH、丙酮酸或没食子酸初始浓度对催化反应的影响,并证明催化剂在催化过程中有较好的稳定性,可使用5次并保持较好的活性.此外,LaTi0.15Cu0.85O3可催化臭氧氧化实际废水中不容易被O3直接氧化的有机污染物〔46〕,LaTi0.15Co0.85O3与LaTi0.15Cu0.85O3还被用于催化臭氧氧化药物分子,包括双氯芬酸(DCF)、合成激素17α-乙炔基二醇(EST)和磺胺甲唑(SMX).在钙钛矿/O3/DCF和钙钛矿/O3/EST体系,除研究反应过程中DCF、EST降解情况和TOC去除率情况外,通过研究反应体系中溶解臭氧的浓度变化发现,臭氧可直接降解DCF和EST及中间产物;但随着反应进行,反应体系产生非臭氧直接降解的过程,可能产生自由基或者其他ROS〔47〕.在钙钛矿/O3/SMX体系,F. Beltrán等〔48〕提出臭氧氧化过程的动力学. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Perovskite catalytic ozonation of pyruvic acid in water: Operating conditions influence and kinetics
2
2006
... F. Beltran课题组〔43〕通过溶胶-凝胶法合成不同钙钛矿氧化物LaTi1-xMexO3(Me=Cu,Co),包括LaTi0.15Cu0.85O3、LaTi0.4Cu0.6O3、LaTi0.78Cu0.22O3、LaTi0.15Co0.85O3、LaTi0.4Cu0.6O3,用于催化臭氧氧化丙酮酸.研究发现LaTi1-xMexO3均能提高O3的氧化能力,且LaTi0.15Cu0.85O3具有最好的催化性能.F. Beltran课题组进一步使用LaTi0.15Cu0.85O3钙钛矿催化臭氧氧化丙酮酸〔44〕和没食子酸〔45〕,系统地研究操作条件,包括臭氧浓度、催化剂用量、温度、pH、丙酮酸或没食子酸初始浓度对催化反应的影响,并证明催化剂在催化过程中有较好的稳定性,可使用5次并保持较好的活性.此外,LaTi0.15Cu0.85O3可催化臭氧氧化实际废水中不容易被O3直接氧化的有机污染物〔46〕,LaTi0.15Co0.85O3与LaTi0.15Cu0.85O3还被用于催化臭氧氧化药物分子,包括双氯芬酸(DCF)、合成激素17α-乙炔基二醇(EST)和磺胺甲唑(SMX).在钙钛矿/O3/DCF和钙钛矿/O3/EST体系,除研究反应过程中DCF、EST降解情况和TOC去除率情况外,通过研究反应体系中溶解臭氧的浓度变化发现,臭氧可直接降解DCF和EST及中间产物;但随着反应进行,反应体系产生非臭氧直接降解的过程,可能产生自由基或者其他ROS〔47〕.在钙钛矿/O3/SMX体系,F. Beltrán等〔48〕提出臭氧氧化过程的动力学. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Catalytic ozonation of phenolic compounds: The case of gallic acid
2
2006
... F. Beltran课题组〔43〕通过溶胶-凝胶法合成不同钙钛矿氧化物LaTi1-xMexO3(Me=Cu,Co),包括LaTi0.15Cu0.85O3、LaTi0.4Cu0.6O3、LaTi0.78Cu0.22O3、LaTi0.15Co0.85O3、LaTi0.4Cu0.6O3,用于催化臭氧氧化丙酮酸.研究发现LaTi1-xMexO3均能提高O3的氧化能力,且LaTi0.15Cu0.85O3具有最好的催化性能.F. Beltran课题组进一步使用LaTi0.15Cu0.85O3钙钛矿催化臭氧氧化丙酮酸〔44〕和没食子酸〔45〕,系统地研究操作条件,包括臭氧浓度、催化剂用量、温度、pH、丙酮酸或没食子酸初始浓度对催化反应的影响,并证明催化剂在催化过程中有较好的稳定性,可使用5次并保持较好的活性.此外,LaTi0.15Cu0.85O3可催化臭氧氧化实际废水中不容易被O3直接氧化的有机污染物〔46〕,LaTi0.15Co0.85O3与LaTi0.15Cu0.85O3还被用于催化臭氧氧化药物分子,包括双氯芬酸(DCF)、合成激素17α-乙炔基二醇(EST)和磺胺甲唑(SMX).在钙钛矿/O3/DCF和钙钛矿/O3/EST体系,除研究反应过程中DCF、EST降解情况和TOC去除率情况外,通过研究反应体系中溶解臭氧的浓度变化发现,臭氧可直接降解DCF和EST及中间产物;但随着反应进行,反应体系产生非臭氧直接降解的过程,可能产生自由基或者其他ROS〔47〕.在钙钛矿/O3/SMX体系,F. Beltrán等〔48〕提出臭氧氧化过程的动力学. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Ozonation of phenolic waste-waters in the presence of a perovskite type catalyst
2
2007
... F. Beltran课题组〔43〕通过溶胶-凝胶法合成不同钙钛矿氧化物LaTi1-xMexO3(Me=Cu,Co),包括LaTi0.15Cu0.85O3、LaTi0.4Cu0.6O3、LaTi0.78Cu0.22O3、LaTi0.15Co0.85O3、LaTi0.4Cu0.6O3,用于催化臭氧氧化丙酮酸.研究发现LaTi1-xMexO3均能提高O3的氧化能力,且LaTi0.15Cu0.85O3具有最好的催化性能.F. Beltran课题组进一步使用LaTi0.15Cu0.85O3钙钛矿催化臭氧氧化丙酮酸〔44〕和没食子酸〔45〕,系统地研究操作条件,包括臭氧浓度、催化剂用量、温度、pH、丙酮酸或没食子酸初始浓度对催化反应的影响,并证明催化剂在催化过程中有较好的稳定性,可使用5次并保持较好的活性.此外,LaTi0.15Cu0.85O3可催化臭氧氧化实际废水中不容易被O3直接氧化的有机污染物〔46〕,LaTi0.15Co0.85O3与LaTi0.15Cu0.85O3还被用于催化臭氧氧化药物分子,包括双氯芬酸(DCF)、合成激素17α-乙炔基二醇(EST)和磺胺甲唑(SMX).在钙钛矿/O3/DCF和钙钛矿/O3/EST体系,除研究反应过程中DCF、EST降解情况和TOC去除率情况外,通过研究反应体系中溶解臭氧的浓度变化发现,臭氧可直接降解DCF和EST及中间产物;但随着反应进行,反应体系产生非臭氧直接降解的过程,可能产生自由基或者其他ROS〔47〕.在钙钛矿/O3/SMX体系,F. Beltrán等〔48〕提出臭氧氧化过程的动力学. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Perovskite catalytic ozonation of some pharmaceutical compounds in water
3
2010
... F. Beltran课题组〔43〕通过溶胶-凝胶法合成不同钙钛矿氧化物LaTi1-xMexO3(Me=Cu,Co),包括LaTi0.15Cu0.85O3、LaTi0.4Cu0.6O3、LaTi0.78Cu0.22O3、LaTi0.15Co0.85O3、LaTi0.4Cu0.6O3,用于催化臭氧氧化丙酮酸.研究发现LaTi1-xMexO3均能提高O3的氧化能力,且LaTi0.15Cu0.85O3具有最好的催化性能.F. Beltran课题组进一步使用LaTi0.15Cu0.85O3钙钛矿催化臭氧氧化丙酮酸〔44〕和没食子酸〔45〕,系统地研究操作条件,包括臭氧浓度、催化剂用量、温度、pH、丙酮酸或没食子酸初始浓度对催化反应的影响,并证明催化剂在催化过程中有较好的稳定性,可使用5次并保持较好的活性.此外,LaTi0.15Cu0.85O3可催化臭氧氧化实际废水中不容易被O3直接氧化的有机污染物〔46〕,LaTi0.15Co0.85O3与LaTi0.15Cu0.85O3还被用于催化臭氧氧化药物分子,包括双氯芬酸(DCF)、合成激素17α-乙炔基二醇(EST)和磺胺甲唑(SMX).在钙钛矿/O3/DCF和钙钛矿/O3/EST体系,除研究反应过程中DCF、EST降解情况和TOC去除率情况外,通过研究反应体系中溶解臭氧的浓度变化发现,臭氧可直接降解DCF和EST及中间产物;但随着反应进行,反应体系产生非臭氧直接降解的过程,可能产生自由基或者其他ROS〔47〕.在钙钛矿/O3/SMX体系,F. Beltrán等〔48〕提出臭氧氧化过程的动力学. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
... 〔
47〕
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Catalysts to improve the abatement of sulfamethoxazole and the resulting organic carbon in water during ozonation
3
2009
... F. Beltran课题组〔43〕通过溶胶-凝胶法合成不同钙钛矿氧化物LaTi1-xMexO3(Me=Cu,Co),包括LaTi0.15Cu0.85O3、LaTi0.4Cu0.6O3、LaTi0.78Cu0.22O3、LaTi0.15Co0.85O3、LaTi0.4Cu0.6O3,用于催化臭氧氧化丙酮酸.研究发现LaTi1-xMexO3均能提高O3的氧化能力,且LaTi0.15Cu0.85O3具有最好的催化性能.F. Beltran课题组进一步使用LaTi0.15Cu0.85O3钙钛矿催化臭氧氧化丙酮酸〔44〕和没食子酸〔45〕,系统地研究操作条件,包括臭氧浓度、催化剂用量、温度、pH、丙酮酸或没食子酸初始浓度对催化反应的影响,并证明催化剂在催化过程中有较好的稳定性,可使用5次并保持较好的活性.此外,LaTi0.15Cu0.85O3可催化臭氧氧化实际废水中不容易被O3直接氧化的有机污染物〔46〕,LaTi0.15Co0.85O3与LaTi0.15Cu0.85O3还被用于催化臭氧氧化药物分子,包括双氯芬酸(DCF)、合成激素17α-乙炔基二醇(EST)和磺胺甲唑(SMX).在钙钛矿/O3/DCF和钙钛矿/O3/EST体系,除研究反应过程中DCF、EST降解情况和TOC去除率情况外,通过研究反应体系中溶解臭氧的浓度变化发现,臭氧可直接降解DCF和EST及中间产物;但随着反应进行,反应体系产生非臭氧直接降解的过程,可能产生自由基或者其他ROS〔47〕.在钙钛矿/O3/SMX体系,F. Beltrán等〔48〕提出臭氧氧化过程的动力学. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
... 〔
48〕
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Enhanced catalytic activity of templated-double perovskite with 3D network structure for salicylic acid degradation under microwave irradiation: Insight into the catalytic mechanism
1
2019
... 镧系元素的原子半径非常有利于ABO3立方晶格的形成〔49〕,且镧系金属阳离子可促进B位金属阳离子在催化反应中的暴露并且不影响催化活性,因此镧系元素(主要是La、Ce、Pr)作为钙钛矿的A位金属受到广泛的研究.C. A. Orge等〔36〕研究不同B位金属的镧基钙钛LaBO3(B=Fe、Ni、Co、Mn)在催化臭氧氧化中的活性.草酸是常见的有机物氧化中间体之一,臭氧很难将其降解,C. A. Orge等发现,LaBO3(B=Fe、Ni、Co、Mn)能促进臭氧对草酸的降解,其催化活性LaMnO3 > LaCoO3 > LaNiO3 > LaFeO3 > 仅臭氧;通过对LaMnO3/O3/草酸体系进一步研究发现,除溶液相中的·OH外,表面反应在催化臭氧降解草酸过程中也发挥重要的作用.更进一步,虽然LaCoO3的BET比表面积比LaMnO3低一倍,但两者几乎有同样的催化效果,这得益于LaCoO3表面存在大量的氧空位,从而证实氧空位对催化活性的重要作用. ...
Catalytic ozonation in water: Controversies and questions. Discussion pape
1
2013
... 在非均相催化臭氧氧化反应中,臭氧吸附在催化剂表面或者有机污染物吸附在催化剂表面或者两者都吸附在催化剂表面是催化剂发挥作用必备的条件〔50〕.增大催化剂的比表面积使更多的活性位点暴露可提高催化活性.S. Afzal等〔51〕以SBA-15为模板,通过纳米浇铸技术合成介孔纳米浇铸钙钛矿NC-LaMnO3与NC-LaFeO3,与溶胶-凝胶法制备的未浇铸钙钛矿LaMnO3和LaFeO3相比,纳米浇铸的钙钛矿具有更高的比表面积和更大的孔径,且在催化臭氧氧化2-氯苯酚时有更好的去除效果及TOC去除率. ...
High surface area mesoporous nanocast LaMO3(M=Mn, Fe)perovskites for efficient catalytic ozonation and an insight into probable catalytic mechanism
4
2017
... 在非均相催化臭氧氧化反应中,臭氧吸附在催化剂表面或者有机污染物吸附在催化剂表面或者两者都吸附在催化剂表面是催化剂发挥作用必备的条件〔50〕.增大催化剂的比表面积使更多的活性位点暴露可提高催化活性.S. Afzal等〔51〕以SBA-15为模板,通过纳米浇铸技术合成介孔纳米浇铸钙钛矿NC-LaMnO3与NC-LaFeO3,与溶胶-凝胶法制备的未浇铸钙钛矿LaMnO3和LaFeO3相比,纳米浇铸的钙钛矿具有更高的比表面积和更大的孔径,且在催化臭氧氧化2-氯苯酚时有更好的去除效果及TOC去除率. ...
... 在非均相催化臭氧氧化过程中,以自由基为基础的氧化反应是目前公认的降解有机污染物的有效途径之一,降解机理如图 4所示〔51〕. ...
... 在探究非均相催化臭氧氧化的机理之前,确认自由基是否参与反应及在整个催化臭氧氧化过程中自由基的种类和贡献至关重要.S. Afzal等〔51〕证实·OH是NC-LaMnO3/O3/2-氯苯酚体系中主要的活性氧物种,且·OH的主要作用是使中间体矿化,2-氯苯酚可被O3直接氧化降解.更进一步,催化剂表面更多的路易斯酸性位点可吸附更多的水分子生成更多的表面羟基,从而与臭氧相互作用产生更多的·OH.该研究还发现,相比于去质子化(MnO-)的表面羟基,质子化(MnOH2+)和中性(MnOH)的表面羟基具有更高的催化活性.除此之外,Mn3+-Mn4+氧化还原循环过程中电子传递可促进O3分解产生更多·OH,反应过程中产生的H2O2也会促进有机污染物的氧化〔52〕. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Mechanism of catalytic ozonation in Fe2O3/Al2O3@SBA-15 aqueous suspension for destruction of ibuprofen
1
2015
... 在探究非均相催化臭氧氧化的机理之前,确认自由基是否参与反应及在整个催化臭氧氧化过程中自由基的种类和贡献至关重要.S. Afzal等〔51〕证实·OH是NC-LaMnO3/O3/2-氯苯酚体系中主要的活性氧物种,且·OH的主要作用是使中间体矿化,2-氯苯酚可被O3直接氧化降解.更进一步,催化剂表面更多的路易斯酸性位点可吸附更多的水分子生成更多的表面羟基,从而与臭氧相互作用产生更多的·OH.该研究还发现,相比于去质子化(MnO-)的表面羟基,质子化(MnOH2+)和中性(MnOH)的表面羟基具有更高的催化活性.除此之外,Mn3+-Mn4+氧化还原循环过程中电子传递可促进O3分解产生更多·OH,反应过程中产生的H2O2也会促进有机污染物的氧化〔52〕. ...
Tailored synthesis of active reduced graphene oxides from waste graphite: Structural defects and pollutant-dependent reactive radicals in aqueous organics decontamination
1
2018
... 通过相同或不同价态的其他阳离子C或D部分取代A位或B位元素可形成取代型的钙钛矿A1-xCxB1-yDyO3(0≤x≤1),可能会改变B位金属氧化态或造成结构缺陷,比如氧空位;除此之外,部分取代B位金属还可提高钙钛矿的稳定性和催化活性〔42〕.在一些非金属材料如石墨烯〔53〕、石墨相氮化碳(g-C3N4)〔54〕的研究中也报道结构缺陷有促进臭氧分解的作用.研究证实提高La1-xCaxCoO3钙钛矿中Ca掺杂量会导致表面氧空位出现〔42〕.少量Ni取代B位金属Fe生成的LaFe0.95Ni0.05O3比LaFeO3有更高的催化臭氧分解的能力〔55〕.C. A. Orge使用Cu部分取代B位金属Fe合成LaFe0.7Cu0.3O3和LaFe0.9Cu0.1O3钙钛矿,其催化臭氧氧化草酸的活性较未取代的LaFeO3明显降低;但通过Cu部分取代A位La元素合成的La0.8Ce0.2Al0.7Cu0.3在催化臭氧氧化草酸的研究中取得比LaAl0.7Cu0.3更好的效果.该工作总结得出,催化剂表面不仅需要臭氧的吸附位点,比如氧空位,B位金属对氧化能力的调节也很重要,所以还需要更深入的研究. ...
Synthesis of N-doped graphene by chemical vapor deposition and its electrical properties
1
2009
... 通过相同或不同价态的其他阳离子C或D部分取代A位或B位元素可形成取代型的钙钛矿A1-xCxB1-yDyO3(0≤x≤1),可能会改变B位金属氧化态或造成结构缺陷,比如氧空位;除此之外,部分取代B位金属还可提高钙钛矿的稳定性和催化活性〔42〕.在一些非金属材料如石墨烯〔53〕、石墨相氮化碳(g-C3N4)〔54〕的研究中也报道结构缺陷有促进臭氧分解的作用.研究证实提高La1-xCaxCoO3钙钛矿中Ca掺杂量会导致表面氧空位出现〔42〕.少量Ni取代B位金属Fe生成的LaFe0.95Ni0.05O3比LaFeO3有更高的催化臭氧分解的能力〔55〕.C. A. Orge使用Cu部分取代B位金属Fe合成LaFe0.7Cu0.3O3和LaFe0.9Cu0.1O3钙钛矿,其催化臭氧氧化草酸的活性较未取代的LaFeO3明显降低;但通过Cu部分取代A位La元素合成的La0.8Ce0.2Al0.7Cu0.3在催化臭氧氧化草酸的研究中取得比LaAl0.7Cu0.3更好的效果.该工作总结得出,催化剂表面不仅需要臭氧的吸附位点,比如氧空位,B位金属对氧化能力的调节也很重要,所以还需要更深入的研究. ...
Highly active and humidity resistive perovskite LaFeO3 based catalysts for efficient ozone decomposition
1
2019
... 通过相同或不同价态的其他阳离子C或D部分取代A位或B位元素可形成取代型的钙钛矿A1-xCxB1-yDyO3(0≤x≤1),可能会改变B位金属氧化态或造成结构缺陷,比如氧空位;除此之外,部分取代B位金属还可提高钙钛矿的稳定性和催化活性〔42〕.在一些非金属材料如石墨烯〔53〕、石墨相氮化碳(g-C3N4)〔54〕的研究中也报道结构缺陷有促进臭氧分解的作用.研究证实提高La1-xCaxCoO3钙钛矿中Ca掺杂量会导致表面氧空位出现〔42〕.少量Ni取代B位金属Fe生成的LaFe0.95Ni0.05O3比LaFeO3有更高的催化臭氧分解的能力〔55〕.C. A. Orge使用Cu部分取代B位金属Fe合成LaFe0.7Cu0.3O3和LaFe0.9Cu0.1O3钙钛矿,其催化臭氧氧化草酸的活性较未取代的LaFeO3明显降低;但通过Cu部分取代A位La元素合成的La0.8Ce0.2Al0.7Cu0.3在催化臭氧氧化草酸的研究中取得比LaAl0.7Cu0.3更好的效果.该工作总结得出,催化剂表面不仅需要臭氧的吸附位点,比如氧空位,B位金属对氧化能力的调节也很重要,所以还需要更深入的研究. ...
Catalytic ozonation of oxalic acid in an aqueous TiO2 slurry reactor
1
2002
... TiO2可有效催化臭氧氧化草酸〔56〕、药物分子(萘普生和卡马西平)〔57〕、苯酚〔58〕等,SrTiO3钙钛矿的基本骨架为Ti-O多面体,呈现出类似TiO2的能带结构,从而产生类似的半导体性质.J. J. Wu等〔59〕将SrTiO3用于催化臭氧氧化草酸,SrTiO3可使草酸的去除率提高36.5%,且可稳定循环使用4次,其结构和催化活性不发生改变. ...
Catalytic ozonation of naproxen and carbamazepine on titanium dioxide
1
2008
... TiO2可有效催化臭氧氧化草酸〔56〕、药物分子(萘普生和卡马西平)〔57〕、苯酚〔58〕等,SrTiO3钙钛矿的基本骨架为Ti-O多面体,呈现出类似TiO2的能带结构,从而产生类似的半导体性质.J. J. Wu等〔59〕将SrTiO3用于催化臭氧氧化草酸,SrTiO3可使草酸的去除率提高36.5%,且可稳定循环使用4次,其结构和催化活性不发生改变. ...
Ozonolysis of phenols in aqueous solution
1
2003
... TiO2可有效催化臭氧氧化草酸〔56〕、药物分子(萘普生和卡马西平)〔57〕、苯酚〔58〕等,SrTiO3钙钛矿的基本骨架为Ti-O多面体,呈现出类似TiO2的能带结构,从而产生类似的半导体性质.J. J. Wu等〔59〕将SrTiO3用于催化臭氧氧化草酸,SrTiO3可使草酸的去除率提高36.5%,且可稳定循环使用4次,其结构和催化活性不发生改变. ...
Catalytic ozonation of oxalic acid using SrTiO3 catalyst
2
2011
... TiO2可有效催化臭氧氧化草酸〔56〕、药物分子(萘普生和卡马西平)〔57〕、苯酚〔58〕等,SrTiO3钙钛矿的基本骨架为Ti-O多面体,呈现出类似TiO2的能带结构,从而产生类似的半导体性质.J. J. Wu等〔59〕将SrTiO3用于催化臭氧氧化草酸,SrTiO3可使草酸的去除率提高36.5%,且可稳定循环使用4次,其结构和催化活性不发生改变. ...
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Synchronously degradation benzotriazole and elimination bromate by perovskite oxides catalytic ozonation: Performance and reaction mechanism
1
2018
... Perovskites and their catalytic performance
Table 3 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| LaCoO3 | 苯并三唑(BZA) | BZA:100%(CO或SO,15 min);TOC:67%(CO,60 min),60%(SO,60 min) | O3 2 mg/L;BZA 10 mg/L;催化剂0.5 g/L | 表面羟基,Co2+/Co3+加速O3分解 | 〔60〕 |
| NC-LaMnO3 | 2-氯苯酚(2-CP) | TOC:80%(CO,75 min),25%(SO,75 min) | O3 20 mg/L,4 mg/min;2-CP 50 mg/L;催化剂0.3 g/L;pH 5.56 | 表面羟基(pH=或 < pHpzc时活性最高)为O3提供吸附位点,Mn3+/Mn4+之间的电子传递驱动O3分解产生ROS | 〔51〕 |
| NC-LaCoO3 | 2-氯苯酚(2-CP) | TOC:68%(CO,75 min),25%(SO,75 min) | | — | |
| LaMn4Ox | 草酸(OA);苯并三唑(BTA) | OA:100%(CO,45 min),10%(SO,45 min);BTA:100%(CO,30 min),100%(SO,60 min);TOC:95%(CO,90 min),50%(SO,90 min) | O3 20 mg/L,100 mL/min;OA 50 mg/L;BTA 50 mg/L;催化剂0.2 g/L;pH 6.8 | 表面氧空位,表面羟基及Lewis酸性位点 | 〔37〕 |
| SrTiO3 | 草酸(OA) | OA:45.8%(CO,60 min),9.3%(SO,60 min) | O3:20 mg/(L·min);OA 100 mg/L;催化剂1.25 g/L;pH 3 | 未探究 | 〔59〕 |
| LaTi0.15Cu0.85O3 | 丙酮酸 | 丙酮酸:75%(CO,200 min),5%(SO,200 min) | O3 50 mg/L,40 L/h;丙酮酸5 mmol/L;catalyst 1 g/L;pH 6 | 未探究 | 〔43, 44〕 |
| LaTi0.15Cu0.85O3 | 没食子酸 | 没食子酸:100%(CO,120 min),100%(SO,180 min);TOC:50%(CO,180 min),35%(SO,180 min) | O3 5 mg/L,40 L/h;没食子酸1 mmol/L;催化剂1 g/L;pH 3.5 | 未探究 | 〔45〕 |
| LaTi0.15Cu0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),85%(CO,2 h) | O3 20 mg/L,40 L/h;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:95%(CO,120 min),40%(SO,120 min);EST-TOC:90%(CO,120 min),65%(SO,120 min) | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Cu0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | COD:40%(SO,320 min),70%(CO,320 min) | O3 50 mg/L,40 L/h;COD 1 100 mg/L;催化剂1 g/L;pH 5.8 | 未探究 | 〔46〕 |
| LaTi0.15Co0.85O3 | 双氯芬酸(DCF);17α-乙炔基二醇(EST) | DCF:100%(CO或SO,10 min);TOC:78%(CO,120 min),40%(SO,120 min);EST-TOC:80%(CO,120 min),65%(SO,120 min); | O3 20 mg/L;EST 9 mg/L;DCF 30 mg/L;催化剂0.1 g/L;pH 7 | 未探究 | 〔47〕 |
| LaTi0.15Co0.85O3 | 磺胺甲唑(SMX) | SMX:100%(CO或SO,10 min);TOC:28%(SO,2 h),60%(CO,2 h) | O3 20 mg/L;SMX 30 mg/L;催化剂1 g/L;pH 7;TOC 15 mg/L | 未探究 | 〔48〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Enhanced catalytic ozonation treatment of dibutyl phthalate enabled by porous magnetic Ag-doped ferrospinel MnFe2O4 materials: Performance and mechanism
4
2018
... 尖晶石多金属氧化物与钙钛矿氧化物一样,具有热力学稳定的结构,可变的组成,丰富的表面羟基,在催化臭氧氧化体系有着广泛的应用.尖晶石多金属氧化物通式为AB2O4(A和B为过渡金属离子)〔61〕,在应用于催化臭氧氧化体系中的尖晶石中,较常见的B位金属有Fe3+、Mn3+和Al3+,常见的A位金属主要有Mn2+、Ni2+、Co2+、Cu2+和Zn2+等.研究证明A位和B位金属具有协同机制〔62〕. ...
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
... 通过掺杂金属可显著提高尖晶石的催化活性.Zimeng Wang等〔61〕通过溶胶-凝胶法制备MnFe2O4尖晶石铁氧体并把Ag掺杂其中,研究对比MnFe2O4和Ag-MnFe2O4催化臭氧氧化邻苯二甲酸二丁酯(DBP)的催化活性.通过表征发现Ag的掺杂会提高MnFe2O4的BET比表面积及孔径;FTIR显示,Ag掺杂后催化剂表面的路易斯酸性位点增加,可促进形成更多的表面羟基,从而促进臭氧与催化剂表面的相互作用,产生更多的ROS;数据显示掺杂质量分数0.5%的Ag使表面羟基的密度提高了2.5倍,使MnFe2O4的催化活性提高了40%.除此之外,循环伏安实验(CV)证明适量的Ag掺杂可降低Ag-MnFe2O4的还原电势,因而降低初始氧化还原反应的活化能垒;XPS数据进一步显示Ag还参与催化臭氧氧化反应,促进Mn2+/Mn3+与O3之间的电子传递.该研究提出其可能的反应机理,如式(8)~式(15)及图 10所示. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Sulfate radical-mediated degradation of sulfadiazine by CuFeO2 rhombohedral crystal-catalyzed peroxymonosulfate: Synergistic effects and mechanisms
1
2016
... 尖晶石多金属氧化物与钙钛矿氧化物一样,具有热力学稳定的结构,可变的组成,丰富的表面羟基,在催化臭氧氧化体系有着广泛的应用.尖晶石多金属氧化物通式为AB2O4(A和B为过渡金属离子)〔61〕,在应用于催化臭氧氧化体系中的尖晶石中,较常见的B位金属有Fe3+、Mn3+和Al3+,常见的A位金属主要有Mn2+、Ni2+、Co2+、Cu2+和Zn2+等.研究证明A位和B位金属具有协同机制〔62〕. ...
Spinel ferrite magnetic adsorbents: Alternative future materials for water purification?
2
2016
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
... 尖晶石铁氧体MFe2O4(M=Mn2+、Ni2+、Co2+、Cu2+、Zn2+)是催化臭氧氧化体系应用最广泛也是研究最多的尖晶石氧化物,具有高密度的表面羟基(Me-OH)〔69〕,大量的氧空位,有丰富且廉价的原料来源〔63〕.除此之外尖晶石铁氧体还具有磁性,在完成催化反应后,可通过磁吸工艺把磁性颗粒从溶液中分离出来〔69〕.Hui Zhao等〔20〕以水热法合成具有磁性的NiFe2O4尖晶石铁氧体,将其用于催化臭氧氧化苯酚,并利用其自带的顺磁性成功分离纳米颗粒与处理后的废水溶液. ...
Magnetically separable and durable MnFe2O4 for efficient catalytic ozonation of organic pollutants
3
2014
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
... 锰的氧化物是催化臭氧氧化体系研究最广泛的催化剂,臭氧在其催化下具有较高分解速率〔72〕.在O3氧化4-氯苯酚(4-CP)体系,MnFe2O4的加入显著提高体系的Rct值(),说明MnFe2O4促进O3分解同时促进·OH的产生;通过对比MnFe2O4与锰铁单金属氧化物(Mn3O4和Fe2O3)混合催化剂[其中n(Mn)/n(Fe)=1:2,与MnFe2O4中两种金属比例相同]催化O3氧化4-CP体系TOC去除率发现,MnFe2O4有更好的催化活性,说明Mn与Fe具有很好的协同效应〔64〕. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Catalytic ozonation of N, N-dimethylacetamide(DMAC) in aqueous solution using nanoscaled magnetic CuFe2O4
2
2018
... 除NiFe2O4外,CuFe2O4也被广泛应用于催化臭氧氧化中.Fei Qi等〔70〕在CuFe2O4催化臭氧氧化非那西丁降解的研究中发现,尽管O3分子可很快与非那西丁反应,但不能有效地使其矿化;而加入CuFe2O4后非那西丁矿化率提高了一倍.研究还发现加入CuFe2O4有2个作用,一是促进臭氧在气相与液相之间的传质及利用,二是促进·OH的产生.之后FeiQi等〔71〕更进一步地研究非那西丁降解的中间体,发现催化剂表面的·OH(吸附·OH)的原位氧化导致中间体的矿化.此外,Heng Zhang等〔65〕发现CuFe2O4/O3体系也可用于N,N-二甲基乙酰胺(DMAC)的降解,使用CuFe2O4/O3处理后DMAC的生物可降解性(BOD5/COD)有所提高,且同样证实羟基自由基在催化氧化中的作用,并认为有机物被·OH氧化的过程发生在催化剂表面附近. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Effective mineralization of quinoline and bio-treated coking wastewater by catalytic ozonation using CuFe2O4/Sepiolite catalyst: Efficiency and mechanism
2
2019
... 不过尖晶石氧化物催化臭氧氧化体系中涉及的ROS也始终存在争议.Dan Liu等〔66〕将CuFe2O4尖晶石铁氧体负载在海泡石上制备CuFe2O4/SEP催化剂,用于催化臭氧氧化喹啉.研究发现除·OH外,O2·- 也是降解喹啉的ROS,且猝灭羟基自由基及清除臭氧,超氧自由基的产生也会被抑制.除此之外,该研究还认为H2O2也是反应体系产生的一种活性含氧物种,是臭氧或羟基自由基与不饱和有机物反应的产物之一,其分解有利于·OH和O2·-的产生;而加入CuFe2O4/SEP催化剂可促进H2O2分解,从而促进有机物的矿化.研究提出的反应机理如图 9所示. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Role of Mg in mesoporous MgFe2O4 for efficient catalytic ozonation of Acid Orange Ⅱ
1
2016
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
Catalytic ozonation for the degradation of 5-sulfosalicylic acid with spinel-type ZnAl2O4 prepared by hydrothermal, sol-gel, and coprecipitation methods: A Comparison Study
3
2018
... 尖晶石的A位与B位金属位于两个不同的晶体学位置上,根据与氧的配位数,A位金属具有四面体氧配位结构,B位金属具有八面体氧配位结构.金属离子的半径和价态、温度、静电能均会影响离子的分布〔63〕.图 6展示代表性的尖晶石晶体结构示意图.常见尖晶石制备方法主要有共沉淀煅烧法〔37〕、溶胶-凝胶法〔17, 61, 64-67〕、水热法〔68〕等. ...
... ZnAl2O4是一种无毒且廉价的尖晶石催化剂,具有良好的扩散性和热稳定性.这些特性使其非常适用于催化臭氧氧化体系.Qizhou Dai等〔68〕分别通过水热法、溶胶-凝胶法及共沉淀法合成ZnAl2O4尖晶石用于催化臭氧氧化5-磺基水杨酸.通过三种方法制备的ZnAl2O4均能促进对5-磺基水杨酸的氧化及对COD的去除,且ZnAl2O4的催化活性与表面羟基密度呈正相关,水热法制备的ZnAl2O4有最高的表面吸附氧含量,相应地也具有最好的催化活性. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Heterogeneous catalytic ozonation of phenacetin in water using magnetic spinel ferrite as catalyst: Comparison of surface property and efficiency
2
2015
... 尖晶石铁氧体MFe2O4(M=Mn2+、Ni2+、Co2+、Cu2+、Zn2+)是催化臭氧氧化体系应用最广泛也是研究最多的尖晶石氧化物,具有高密度的表面羟基(Me-OH)〔69〕,大量的氧空位,有丰富且廉价的原料来源〔63〕.除此之外尖晶石铁氧体还具有磁性,在完成催化反应后,可通过磁吸工艺把磁性颗粒从溶液中分离出来〔69〕.Hui Zhao等〔20〕以水热法合成具有磁性的NiFe2O4尖晶石铁氧体,将其用于催化臭氧氧化苯酚,并利用其自带的顺磁性成功分离纳米颗粒与处理后的废水溶液. ...
... 〔69〕.Hui Zhao等〔20〕以水热法合成具有磁性的NiFe2O4尖晶石铁氧体,将其用于催化臭氧氧化苯酚,并利用其自带的顺磁性成功分离纳米颗粒与处理后的废水溶液. ...
Ozonation of phenacetin in associated with a magnetic catalyst CuFe2O4: The reaction and transformation
2
2015
... 除NiFe2O4外,CuFe2O4也被广泛应用于催化臭氧氧化中.Fei Qi等〔70〕在CuFe2O4催化臭氧氧化非那西丁降解的研究中发现,尽管O3分子可很快与非那西丁反应,但不能有效地使其矿化;而加入CuFe2O4后非那西丁矿化率提高了一倍.研究还发现加入CuFe2O4有2个作用,一是促进臭氧在气相与液相之间的传质及利用,二是促进·OH的产生.之后FeiQi等〔71〕更进一步地研究非那西丁降解的中间体,发现催化剂表面的·OH(吸附·OH)的原位氧化导致中间体的矿化.此外,Heng Zhang等〔65〕发现CuFe2O4/O3体系也可用于N,N-二甲基乙酰胺(DMAC)的降解,使用CuFe2O4/O3处理后DMAC的生物可降解性(BOD5/COD)有所提高,且同样证实羟基自由基在催化氧化中的作用,并认为有机物被·OH氧化的过程发生在催化剂表面附近. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Comparison of phenacetin degradation in aqueous solutions by catalytic ozonation with CuFe2O4 and its precursor: Surface properties, intermediates and reaction mechanisms
1
2016
... 除NiFe2O4外,CuFe2O4也被广泛应用于催化臭氧氧化中.Fei Qi等〔70〕在CuFe2O4催化臭氧氧化非那西丁降解的研究中发现,尽管O3分子可很快与非那西丁反应,但不能有效地使其矿化;而加入CuFe2O4后非那西丁矿化率提高了一倍.研究还发现加入CuFe2O4有2个作用,一是促进臭氧在气相与液相之间的传质及利用,二是促进·OH的产生.之后FeiQi等〔71〕更进一步地研究非那西丁降解的中间体,发现催化剂表面的·OH(吸附·OH)的原位氧化导致中间体的矿化.此外,Heng Zhang等〔65〕发现CuFe2O4/O3体系也可用于N,N-二甲基乙酰胺(DMAC)的降解,使用CuFe2O4/O3处理后DMAC的生物可降解性(BOD5/COD)有所提高,且同样证实羟基自由基在催化氧化中的作用,并认为有机物被·OH氧化的过程发生在催化剂表面附近. ...
VOC removal by synergic effect of combustion catalyst and ozone
1
1996
... 锰的氧化物是催化臭氧氧化体系研究最广泛的催化剂,臭氧在其催化下具有较高分解速率〔72〕.在O3氧化4-氯苯酚(4-CP)体系,MnFe2O4的加入显著提高体系的Rct值(),说明MnFe2O4促进O3分解同时促进·OH的产生;通过对比MnFe2O4与锰铁单金属氧化物(Mn3O4和Fe2O3)混合催化剂[其中n(Mn)/n(Fe)=1:2,与MnFe2O4中两种金属比例相同]催化O3氧化4-CP体系TOC去除率发现,MnFe2O4有更好的催化活性,说明Mn与Fe具有很好的协同效应〔64〕. ...
Ozone decomposition, benzene and CO oxidation over NiMnO3-ilmenite and NiMn2O4-spinel catalysts
1
2001
... 尖晶石锰氧体也是常用的尖晶石之一.D.Mehandjiev等〔73〕对比了NiMnO3与NiMn2O4催化臭氧分解及催化臭氧氧化苯的活性.研究发现NiMnO3与NiMn2O4均有较高的活性,相比之下,NiMnO3表面的氧物种活性更高.Wenkai Zhao等〔74〕通过水热法合成CoMn2O4尖晶石,发现在CO还原气氛中煅烧可增加CoMn2O4表面氧空位及表面羟基密度;实验证明在还原气氛下煅烧的CoMn2O4相较于在空气中煅烧的尖晶石具有更高的催化活性.除表面Lewis酸性位点,Wenkai Zhao等还通过一系列实验证明氧空位可促进吸附的水分子与CoMn2O4相互作用形成表面羟基,从而进一步促进·OH的产生. ...
Enhanced catalytic ozonation over reduced spinel CoMn2O4 for NOx removal: Active site and mechanism analysis
2
2016
... 尖晶石锰氧体也是常用的尖晶石之一.D.Mehandjiev等〔73〕对比了NiMnO3与NiMn2O4催化臭氧分解及催化臭氧氧化苯的活性.研究发现NiMnO3与NiMn2O4均有较高的活性,相比之下,NiMnO3表面的氧物种活性更高.Wenkai Zhao等〔74〕通过水热法合成CoMn2O4尖晶石,发现在CO还原气氛中煅烧可增加CoMn2O4表面氧空位及表面羟基密度;实验证明在还原气氛下煅烧的CoMn2O4相较于在空气中煅烧的尖晶石具有更高的催化活性.除表面Lewis酸性位点,Wenkai Zhao等还通过一系列实验证明氧空位可促进吸附的水分子与CoMn2O4相互作用形成表面羟基,从而进一步促进·OH的产生. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Catalytic ozonation for degradation of sulfamethazine using NiCo2O4 as catalyst
2
2021
... Ni2+/Ni3+与Co2+/Co3+是常见的氧化还原循环,Hai Chen等〔75〕首次合成NiCo2O4尖晶石并用于催化臭氧氧化体系.由于O3可选择性地攻击磺胺二甲嘧啶(SMT)中H-N键和苯环,所以臭氧可快速降解SMT,但无法完全矿化SMT,而在NiCo2O4尖晶石催化下可使TOC去除率提高3倍.该研究认为Ni和Co均在矿化过程中发挥作用,Co不直接参与氧化还原循环,而是作为Lewis酸性位点与吸附水分子作用产生表面羟基形成Ni(Ⅱ)Co2O4-O-H-O3电子传递桥梁,Ni2+用于提供电子使H-O和O-O键断裂产生HO2·和O2·-,与O3反应进一步产生·OH.除晶格氧之外〔14〕,该研究还发现产生的O2·-可还原Ni3+为Ni2+,保证多金属氧化物的持续催化能力. ...
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Mechanism and kinetics of catalytic ozonation for elimination of organic compounds with spinel-type CuAl2O4 and its precursor
1
2019
... Spinel oxides and their catalytic performance
Table 4 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| NiFe2O4 | 苯酚 | 苯酚:55.2%(CO,60 min),38.9%(SO,60 min) | O3 0.75 mg/min;苯酚: 300 mg/L;催化剂1 g/L;pH 6.5±0.1 | Lewis酸性位点 | 〔20〕 |
| NiFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:100%(CO,60 min),42%(SO,60 min) | O3 5 mg/L;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.7 | 表面羟基 | 〔14〕 |
| NiFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1)mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| CuFe2O4 | 非那西丁(PNT) | PNT:100%(CO,15 min),95%(SO,30 min);TOC:50%(CO,130 min),15%(SO,130 min) | O3 0.36 mg/L;PNT 20 mmol/L;催化剂2 g/L;pH 7.72 | - | 〔70〕 |
| CuFe2O4 | N,N-二甲基乙酰胺(DMAC) | DMAC:95.4%(CO,120 min),55.4%(SO,120 min);COD:30.1%(CO),11.1%(SO,120 min);TOC:22.3%(CO,120 min),6.8%(SO,120 min) | O3 0.6 L/min;DMAC 200 mg/L;催化剂30 g/L;pH 6.7 | 表面羟基 | 〔65〕 |
| CuFe2O4 | 草酸(OA) | OA:15%(CO,120 min),4.7%(SO,120 min) | O3(14±1) mg/L,1 L/min;OA5 mmol/L;催化剂1 g/L;pH 2.3±0.02 | 表面羟基 | 〔17〕 |
| ZnFe2O4 | 草酸(OA) | OA:8.5%(CO,120 min),4.7%(SO,120 min) | | | 〔17〕 |
| CoFe2O4 | 草酸(OA) | OA:68.3%(CO,120 min);4.7%(SO,120 min) | | | 〔17〕 |
| CuFe2O4/SEP | 喹啉 | 喹啉:93.3%(SO);100%(CO);TOC:16.8%(SO);90.3%(CO) | O3 1 L/min;喹啉50 mg/L;催化剂1.0 g/L;pH 6.8 | 表面羟基(pH>=pHpzc时比pH < pHpzc更利于催化反应) | 〔66〕 |
| MnFe2O4 | 4-氯苯酚(4-CP) | 4-CP:95.7%(CO,30 min),77%(SO,30 min);TOC:60.3%(CO,90 min),26.8%(SO,90 min) | O3 5 mg/L,1 L/min;4-CP 100 mg/L;催化剂1.0 g/L | 表面羟基(pH>= pHpzc时活性最高),Mn3+/Mn2+、Fe3+/Fe2+、Mn3+/Fe2+促进O3分解 | 〔64〕 |
| Ag-MnFe2O4 | 邻苯二甲酸二丁酯(DBP) | DBP:75%(CO,60 min),30%(SO,60 min) | O3 0.68 mg/min;DBP 0.5 mg/L;催化剂0.01 g/L;pH 7.3 | 表面羟基 | 〔61〕 |
| ZnAl2O4 | 5-磺基水杨酸(SSal) | SSal:64.8%(CO),49.4%(SO);COD:46.2%(CO),33.2%(SO) | O3 5 mg/min;SSal 500 mg/L;催化剂0.2 g/L;pH 7.0 | 表面羟基 | 〔68〕 |
| CoMn2O4 | NOx | NO只能被O3氧化成NO2,·OH可氧化NO与NO2至HNO3 | O3 2.8 mg/L,O3 100 mL/min;NOx 450 mg/L;催化剂0.2 g/L | 氧空位促进H2O的吸附并作用产生表面羟基,促进O3分解 | 〔74〕 |
| CuAl2O4 | 酸性橙7(AO7) | AO7:96%(CO),76%(SO);COD:87.2%(CO),40%(SO) | O3 10.06 mg/min,40 L/h;AO7 100 mg/L;催化剂0.5 g/L;pH 6.54 | 表面Lewis酸性位点(≡Al3+)与H2O作用产生表面羟基,Cu2+/Cu+氧化还原过程促进O3分解 | 〔76〕 |
| NiCo2O4 | 磺胺二甲嘧啶(SMT) | TOC:34.1%(CO),11%(SO) | O3 4.5 mg/min;AO7 20 mg/L;催化剂0.05 g/L;pH 5.2 | Lewis酸性位点,表面羟基 | 〔75〕 |
注: CO是催化臭氧氧化过程, SO是单独臭氧氧化过程. ...
Synthesis and applications of nano-structured iron oxides/hydroxides-a review
1
2010
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
Efficiencies and mechanisms of ZSM5 zeolites loaded with cerium, iron, or manganese oxides for catalytic ozonation of nitrobenzene in water
2
2018
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
... A. Ikhlaq等〔83〕研究ZSM-5沸石催化臭氧降解有机污染物的活性发现,降低SiO2/Al2O3比值更有利于布洛芬在ZSM-5上的吸附,且ZSM-5催化臭氧氧化布洛芬的活性与反应溶液的pH有关,降低pH更有利于对布洛芬的去除.醋酸是催化臭氧氧化布洛芬产生的中间产物,且不容易被臭氧直接氧化,该研究通过实验证明ZSM-5不能促进臭氧氧化降解醋酸,且ZSM-5不能催化溶液中溶解臭氧的分解,因而认为ZSM-5促进吸附在催化剂表面的臭氧和有机污染物的表面反应,只是这不是通过产生自由基进行的.Chunmao Chen等〔78〕将金属(Ce、Fe、Mn)氧化物负载到沸石上,研究其在催化臭氧氧化硝基苯(NB)中的活性.首先,无金属负载的沸石可提高臭氧氧化NB的效率,NaZSM5-38、HZSM5-38、NaZSM5-100可分别去除45.9%、62.3%、59%的TOC,均高于单独臭氧氧化的效果(39.2%),进一步将金属氧化物负载至HZSM5-38表面可提高沸石的催化活性,并且Ce/NaZSM5-38具有最高的催化活性(TOC去除率86.3%).H. Valdés等〔84〕在研究中提出,HCl处理可提高沸石催化臭氧氧化甲基蓝(MB)的效率,通过催化剂表征发现,酸处理降了沸石的pHpzc,增了沸石表面Bronstead酸性位点并且酸处理沸石表面的Bronstead酸性位点是反应的活性位点.在另一项研究中,H. Valdés等〔85〕证实酸处理可提高沸石催化臭氧分解的活性,酸处理使沸石结构中的Al被4个羟基取代(图 12),而臭氧与沸石表面酸性位点的相互作用是催化臭氧分解的关键步骤. ...
Removal mechanism of natural organic matter and organic acid by ozone in the presence of goethite
2
2004
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
... FeOOH及其衍生物在催化臭氧氧化中也展现出较高的催化活性.J. S. Park等〔79, 86〕研究发现针铁矿能显著提高臭氧氧化对氯苯甲酸(p-CBA)和天然有机质(NOM)的效率.针铁矿的主要成分是α-FeOOH,Tao Zhang等〔87〕合成针铁矿(α-FeOOH),并用于催化臭氧氧化4-硝基苯酚(4-NP),α-FeOOH在催化臭氧氧化中的催化活性得到验证,之后研究人员又尝试进一步使用Ce、Co、Mn等元素部分取代Fe形成多金属MexFe1-xOOH以提高其催化活性〔88-90〕. ...
Ozonation catalyzed by the raw bauxite for the degradation of 2, 4, 6-trichloroanisole in drinking water
2
2009
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
... Fei Qi等〔80〕发现使用未加工的铝土矿可显著提高臭氧氧化2,4,6-三氯苯甲醚(TCA)的效率(86%).在此基础上,Fei Qi等〔94〕使用Mn、Fe和Ce对铝土矿进行掺杂并用于催化臭氧氧化2,4,6-三氯苯甲醚(TCA).结果表明,掺杂Mn、Fe和Ce的铝土矿催化TCA降解的活性均有所提高,降解效果Fe-铝土矿 > Mn-铝土矿 > Ce-铝土矿 > 粗铝土矿 > 仅臭氧.金属掺杂提高铝土矿的比表面积、孔体积,也提高表面羟基密度.图 14给出了该研究提出的反应机理:O3和TCA吸附在催化剂表面;O3可直接与TCA反应,微孔表面增加催化剂、污染物和臭氧分子之间碰撞的可能性;O3分解产生·OH使TCA降解,而铝土矿表面的羟基基团是O3与催化剂相互作用的位点. ...
Mechanism of heterogeneous catalytic ozonation of nitrobenzene in aqueous solution with modified ceramic honeycomb
2
2009
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
... 在另一项工作中,Lei Zhao等〔98〕研究反应初始pH对反应体系的影响,发现反应溶液初始pH影响溶液中臭氧浓度及臭氧的利用率;pH在pHpzc附近时,蜂窝陶瓷催化臭氧氧化NB的效率最高,此时表面羟基是电中性的,并且表面羟基密度也具有最高值.除此之外,Lei Zhao等〔81, 99〕在另外的两项工作中还分别研究负载Mn及Cu和Mn的蜂窝陶瓷在催化臭氧氧化NB中的应用,研究发现Mn和Cu的掺杂同样提高催化剂表面的羟基浓度,从而促进·OH的产生,致使蜂窝陶瓷的催化活性有所提高. ...
Pumice-catalyzed ozonation degradation of p-chloronitrobenzene in aqueous solution
3
2012
... 人工合成无机材料作为催化剂及其载体在高级氧化中展现较好应用前景,但其制备成本相对较高,而天然矿物材料来源广泛、价格低廉、制备简单,越来越多的研究专注于天然矿石在高级氧化处理中的应用,尤其是在催化臭氧氧化反应中的应用〔23, 77〕.常用的天然矿石材料有沸石〔78〕、针铁矿〔79〕、铝土矿〔80〕、堇青石〔81〕、浮石〔82〕等. ...
... 浮石(PMC)是一种火山活动中产生的多孔天然材料,它的主要成分是硅、铝、铁的氧化物.Lei Yuan等〔82〕使用天然浮石作为催化臭氧氧化对氯硝基苯(pCNB)的催化剂,通过研究发现,其主要成分是SiO2、Al2O3、Fe2O3、K2O、MgO、Na2O、TiO2,在pCNB降解过程中,·OH是主要的ROS,不带电的表面羟基基团更有利于·OH的产生.在另一项研究中,Guoying Gao等〔100〕使用负载硅酸铁的浮石(FSO/PMC)催化臭氧氧化双氯芬酸(DCF),加入的FSO/PMC促进了O3的传质和溶解,从而促进·OH的产生;除此之外,FSO/PMC促进H2O2的产生与消耗,同样有利于·OH的产生. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Catalytic ozonation for the removal of organic contaminants in water on ZSM-5 zeolites
2
2014
... A. Ikhlaq等〔83〕研究ZSM-5沸石催化臭氧降解有机污染物的活性发现,降低SiO2/Al2O3比值更有利于布洛芬在ZSM-5上的吸附,且ZSM-5催化臭氧氧化布洛芬的活性与反应溶液的pH有关,降低pH更有利于对布洛芬的去除.醋酸是催化臭氧氧化布洛芬产生的中间产物,且不容易被臭氧直接氧化,该研究通过实验证明ZSM-5不能促进臭氧氧化降解醋酸,且ZSM-5不能催化溶液中溶解臭氧的分解,因而认为ZSM-5促进吸附在催化剂表面的臭氧和有机污染物的表面反应,只是这不是通过产生自由基进行的.Chunmao Chen等〔78〕将金属(Ce、Fe、Mn)氧化物负载到沸石上,研究其在催化臭氧氧化硝基苯(NB)中的活性.首先,无金属负载的沸石可提高臭氧氧化NB的效率,NaZSM5-38、HZSM5-38、NaZSM5-100可分别去除45.9%、62.3%、59%的TOC,均高于单独臭氧氧化的效果(39.2%),进一步将金属氧化物负载至HZSM5-38表面可提高沸石的催化活性,并且Ce/NaZSM5-38具有最高的催化活性(TOC去除率86.3%).H. Valdés等〔84〕在研究中提出,HCl处理可提高沸石催化臭氧氧化甲基蓝(MB)的效率,通过催化剂表征发现,酸处理降了沸石的pHpzc,增了沸石表面Bronstead酸性位点并且酸处理沸石表面的Bronstead酸性位点是反应的活性位点.在另一项研究中,H. Valdés等〔85〕证实酸处理可提高沸石催化臭氧分解的活性,酸处理使沸石结构中的Al被4个羟基取代(图 12),而臭氧与沸石表面酸性位点的相互作用是催化臭氧分解的关键步骤. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Role of surface hydroxyl groups of acid-treated natural zeolite on the heterogeneous catalytic ozonation of methylene blue contaminated waters
1
2012
... A. Ikhlaq等〔83〕研究ZSM-5沸石催化臭氧降解有机污染物的活性发现,降低SiO2/Al2O3比值更有利于布洛芬在ZSM-5上的吸附,且ZSM-5催化臭氧氧化布洛芬的活性与反应溶液的pH有关,降低pH更有利于对布洛芬的去除.醋酸是催化臭氧氧化布洛芬产生的中间产物,且不容易被臭氧直接氧化,该研究通过实验证明ZSM-5不能促进臭氧氧化降解醋酸,且ZSM-5不能催化溶液中溶解臭氧的分解,因而认为ZSM-5促进吸附在催化剂表面的臭氧和有机污染物的表面反应,只是这不是通过产生自由基进行的.Chunmao Chen等〔78〕将金属(Ce、Fe、Mn)氧化物负载到沸石上,研究其在催化臭氧氧化硝基苯(NB)中的活性.首先,无金属负载的沸石可提高臭氧氧化NB的效率,NaZSM5-38、HZSM5-38、NaZSM5-100可分别去除45.9%、62.3%、59%的TOC,均高于单独臭氧氧化的效果(39.2%),进一步将金属氧化物负载至HZSM5-38表面可提高沸石的催化活性,并且Ce/NaZSM5-38具有最高的催化活性(TOC去除率86.3%).H. Valdés等〔84〕在研究中提出,HCl处理可提高沸石催化臭氧氧化甲基蓝(MB)的效率,通过催化剂表征发现,酸处理降了沸石的pHpzc,增了沸石表面Bronstead酸性位点并且酸处理沸石表面的Bronstead酸性位点是反应的活性位点.在另一项研究中,H. Valdés等〔85〕证实酸处理可提高沸石催化臭氧分解的活性,酸处理使沸石结构中的Al被4个羟基取代(图 12),而臭氧与沸石表面酸性位点的相互作用是催化臭氧分解的关键步骤. ...
Catalytic ozone aqueous decomposition promoted by natural zeolite and volcanic sand
1
2009
... A. Ikhlaq等〔83〕研究ZSM-5沸石催化臭氧降解有机污染物的活性发现,降低SiO2/Al2O3比值更有利于布洛芬在ZSM-5上的吸附,且ZSM-5催化臭氧氧化布洛芬的活性与反应溶液的pH有关,降低pH更有利于对布洛芬的去除.醋酸是催化臭氧氧化布洛芬产生的中间产物,且不容易被臭氧直接氧化,该研究通过实验证明ZSM-5不能促进臭氧氧化降解醋酸,且ZSM-5不能催化溶液中溶解臭氧的分解,因而认为ZSM-5促进吸附在催化剂表面的臭氧和有机污染物的表面反应,只是这不是通过产生自由基进行的.Chunmao Chen等〔78〕将金属(Ce、Fe、Mn)氧化物负载到沸石上,研究其在催化臭氧氧化硝基苯(NB)中的活性.首先,无金属负载的沸石可提高臭氧氧化NB的效率,NaZSM5-38、HZSM5-38、NaZSM5-100可分别去除45.9%、62.3%、59%的TOC,均高于单独臭氧氧化的效果(39.2%),进一步将金属氧化物负载至HZSM5-38表面可提高沸石的催化活性,并且Ce/NaZSM5-38具有最高的催化活性(TOC去除率86.3%).H. Valdés等〔84〕在研究中提出,HCl处理可提高沸石催化臭氧氧化甲基蓝(MB)的效率,通过催化剂表征发现,酸处理降了沸石的pHpzc,增了沸石表面Bronstead酸性位点并且酸处理沸石表面的Bronstead酸性位点是反应的活性位点.在另一项研究中,H. Valdés等〔85〕证实酸处理可提高沸石催化臭氧分解的活性,酸处理使沸石结构中的Al被4个羟基取代(图 12),而臭氧与沸石表面酸性位点的相互作用是催化臭氧分解的关键步骤. ...
Kinetic decomposition of ozone and para-chlorobenzoic acid(pCBA) during catalytic ozonation
1
2004
... FeOOH及其衍生物在催化臭氧氧化中也展现出较高的催化活性.J. S. Park等〔79, 86〕研究发现针铁矿能显著提高臭氧氧化对氯苯甲酸(p-CBA)和天然有机质(NOM)的效率.针铁矿的主要成分是α-FeOOH,Tao Zhang等〔87〕合成针铁矿(α-FeOOH),并用于催化臭氧氧化4-硝基苯酚(4-NP),α-FeOOH在催化臭氧氧化中的催化活性得到验证,之后研究人员又尝试进一步使用Ce、Co、Mn等元素部分取代Fe形成多金属MexFe1-xOOH以提高其催化活性〔88-90〕. ...
Catalytic ozonation of trace nitrobenzene in water with synthetic goethite
1
2008
... FeOOH及其衍生物在催化臭氧氧化中也展现出较高的催化活性.J. S. Park等〔79, 86〕研究发现针铁矿能显著提高臭氧氧化对氯苯甲酸(p-CBA)和天然有机质(NOM)的效率.针铁矿的主要成分是α-FeOOH,Tao Zhang等〔87〕合成针铁矿(α-FeOOH),并用于催化臭氧氧化4-硝基苯酚(4-NP),α-FeOOH在催化臭氧氧化中的催化活性得到验证,之后研究人员又尝试进一步使用Ce、Co、Mn等元素部分取代Fe形成多金属MexFe1-xOOH以提高其催化活性〔88-90〕. ...
Catalytic ozonation of dimethyl phthalate by Ce-substituted goethite
3
2017
... FeOOH及其衍生物在催化臭氧氧化中也展现出较高的催化活性.J. S. Park等〔79, 86〕研究发现针铁矿能显著提高臭氧氧化对氯苯甲酸(p-CBA)和天然有机质(NOM)的效率.针铁矿的主要成分是α-FeOOH,Tao Zhang等〔87〕合成针铁矿(α-FeOOH),并用于催化臭氧氧化4-硝基苯酚(4-NP),α-FeOOH在催化臭氧氧化中的催化活性得到验证,之后研究人员又尝试进一步使用Ce、Co、Mn等元素部分取代Fe形成多金属MexFe1-xOOH以提高其催化活性〔88-90〕. ...
... Zhiyong Bai等〔88〕使用Ce微量取代Fe合成Ce0.1Fe0.9OOH,用于降解磺胺二甲嘧啶(SMT)、邻苯二甲酸二甲酯(DMP).研究发现,Ce的微量取代可提高针铁矿的比表面积及表面羟基密度,且没有改变其晶体结构,因此,与α-FeOOH相比,Ce0.1Fe0.9OOH催化臭氧氧化DMP的活性更高.除此之外,Ce3+/Ce4+与Fe2+/Fe3+氧化还原循环之间存在的协同作用可增强Ce0.1Fe0.9OOH与O3之间的电子传递能力,从而促进O3分解产生·OH.在催化臭氧氧化SM的研究中,Zhiyong Bai等〔89〕认为Ce0.1Fe0.9OOH可促进O3在气相与液相之间的传质及利用,并提出SMT在Ce0.1Fe0.9OOH催化下被O3降解的路径〔90〕,见图 13. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Catalytic ozonation of sulfa-methazine antibiotics using Ce0.1Fe0.9OOH: Catalyst preparation and performance
2
2016
... Zhiyong Bai等〔88〕使用Ce微量取代Fe合成Ce0.1Fe0.9OOH,用于降解磺胺二甲嘧啶(SMT)、邻苯二甲酸二甲酯(DMP).研究发现,Ce的微量取代可提高针铁矿的比表面积及表面羟基密度,且没有改变其晶体结构,因此,与α-FeOOH相比,Ce0.1Fe0.9OOH催化臭氧氧化DMP的活性更高.除此之外,Ce3+/Ce4+与Fe2+/Fe3+氧化还原循环之间存在的协同作用可增强Ce0.1Fe0.9OOH与O3之间的电子传递能力,从而促进O3分解产生·OH.在催化臭氧氧化SM的研究中,Zhiyong Bai等〔89〕认为Ce0.1Fe0.9OOH可促进O3在气相与液相之间的传质及利用,并提出SMT在Ce0.1Fe0.9OOH催化下被O3降解的路径〔90〕,见图 13. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Catalytic ozonation of sulfa-methazine using Ce0.1Fe0.9OOH as catalyst: Mineralization and catalytic mechanisms
3
2016
... FeOOH及其衍生物在催化臭氧氧化中也展现出较高的催化活性.J. S. Park等〔79, 86〕研究发现针铁矿能显著提高臭氧氧化对氯苯甲酸(p-CBA)和天然有机质(NOM)的效率.针铁矿的主要成分是α-FeOOH,Tao Zhang等〔87〕合成针铁矿(α-FeOOH),并用于催化臭氧氧化4-硝基苯酚(4-NP),α-FeOOH在催化臭氧氧化中的催化活性得到验证,之后研究人员又尝试进一步使用Ce、Co、Mn等元素部分取代Fe形成多金属MexFe1-xOOH以提高其催化活性〔88-90〕. ...
... Zhiyong Bai等〔88〕使用Ce微量取代Fe合成Ce0.1Fe0.9OOH,用于降解磺胺二甲嘧啶(SMT)、邻苯二甲酸二甲酯(DMP).研究发现,Ce的微量取代可提高针铁矿的比表面积及表面羟基密度,且没有改变其晶体结构,因此,与α-FeOOH相比,Ce0.1Fe0.9OOH催化臭氧氧化DMP的活性更高.除此之外,Ce3+/Ce4+与Fe2+/Fe3+氧化还原循环之间存在的协同作用可增强Ce0.1Fe0.9OOH与O3之间的电子传递能力,从而促进O3分解产生·OH.在催化臭氧氧化SM的研究中,Zhiyong Bai等〔89〕认为Ce0.1Fe0.9OOH可促进O3在气相与液相之间的传质及利用,并提出SMT在Ce0.1Fe0.9OOH催化下被O3降解的路径〔90〕,见图 13. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Interface mechanism of catalytic ozonation in an α-Fe0.9Mn0.1OOH aqueous suspension for the removal of iohexol
2
2020
... Pengwei Yan等〔91-92〕也发现Mn部分取代Fe生成的α-Fe0.9Mn0.1OOH催化臭氧氧化碘海醇的活性较α-FeOOH明显提高.Co是催化臭氧氧化中常用的过渡金属元素,Zhenzhen Xu等〔93〕通过共沉淀法合成Co-FeOOH[n(Co)/n(Fe)=0.13]用于催化臭氧氧化阿提洛尔(ATL)——一种可被臭氧直接氧化的药物分子.研究发现,相比于单独臭氧氧化,在CoOOH、FeOOH、Co-FeOOH催化下ATL的伪一级降解速率常数分别提高了2.18、1.6、1.6倍,并且在Co-FeOOH催化下,TOC去除率更高.该研究还发现,ATL及TOC的去除率受反应体系的pH影响较大,因为pH会从以下三个方面影响催化活性:(1)催化剂表面性质,(2)ATL存在形式,(3)O3的自分解程度. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Catalytic ozonation of iohexol with α-Fe0.9Mn0.1OOH in water: Efficiency, degradation mechanism and toxicity evaluation
2
2020
... Pengwei Yan等〔91-92〕也发现Mn部分取代Fe生成的α-Fe0.9Mn0.1OOH催化臭氧氧化碘海醇的活性较α-FeOOH明显提高.Co是催化臭氧氧化中常用的过渡金属元素,Zhenzhen Xu等〔93〕通过共沉淀法合成Co-FeOOH[n(Co)/n(Fe)=0.13]用于催化臭氧氧化阿提洛尔(ATL)——一种可被臭氧直接氧化的药物分子.研究发现,相比于单独臭氧氧化,在CoOOH、FeOOH、Co-FeOOH催化下ATL的伪一级降解速率常数分别提高了2.18、1.6、1.6倍,并且在Co-FeOOH催化下,TOC去除率更高.该研究还发现,ATL及TOC的去除率受反应体系的pH影响较大,因为pH会从以下三个方面影响催化活性:(1)催化剂表面性质,(2)ATL存在形式,(3)O3的自分解程度. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Efficiency and mechanism of atenolol decomposition in Co-FeOOH catalytic ozonation
2
2019
... Pengwei Yan等〔91-92〕也发现Mn部分取代Fe生成的α-Fe0.9Mn0.1OOH催化臭氧氧化碘海醇的活性较α-FeOOH明显提高.Co是催化臭氧氧化中常用的过渡金属元素,Zhenzhen Xu等〔93〕通过共沉淀法合成Co-FeOOH[n(Co)/n(Fe)=0.13]用于催化臭氧氧化阿提洛尔(ATL)——一种可被臭氧直接氧化的药物分子.研究发现,相比于单独臭氧氧化,在CoOOH、FeOOH、Co-FeOOH催化下ATL的伪一级降解速率常数分别提高了2.18、1.6、1.6倍,并且在Co-FeOOH催化下,TOC去除率更高.该研究还发现,ATL及TOC的去除率受反应体系的pH影响较大,因为pH会从以下三个方面影响催化活性:(1)催化剂表面性质,(2)ATL存在形式,(3)O3的自分解程度. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Comparison of the efficiency and mechanism of catalytic ozonation of 2, 4, 6-trichloroanisole by iron and manganese modified bauxite
1
2012
... Fei Qi等〔80〕发现使用未加工的铝土矿可显著提高臭氧氧化2,4,6-三氯苯甲醚(TCA)的效率(86%).在此基础上,Fei Qi等〔94〕使用Mn、Fe和Ce对铝土矿进行掺杂并用于催化臭氧氧化2,4,6-三氯苯甲醚(TCA).结果表明,掺杂Mn、Fe和Ce的铝土矿催化TCA降解的活性均有所提高,降解效果Fe-铝土矿 > Mn-铝土矿 > Ce-铝土矿 > 粗铝土矿 > 仅臭氧.金属掺杂提高铝土矿的比表面积、孔体积,也提高表面羟基密度.图 14给出了该研究提出的反应机理:O3和TCA吸附在催化剂表面;O3可直接与TCA反应,微孔表面增加催化剂、污染物和臭氧分子之间碰撞的可能性;O3分解产生·OH使TCA降解,而铝土矿表面的羟基基团是O3与催化剂相互作用的位点. ...
Degradation of nitroben-zene in aqueous solution by ozone-ceramic honeycomb
1
2005
... 蜂窝陶瓷(Ceramic Honeycomb,CH)是结构像蜂窝的多孔陶瓷,可由堇青石、莫来石、钛酸铝、氧化锆、碳化硅质、堇青-莫来石等多种材料组成,其中最广泛应用的是具有高熔点、高机械强度的堇青石(2 MgO·2Al2O3·5SiO2).堇青石可促进硝基苯(NB)在臭氧体系中的降解(降解率50%)及体系TOC的去除(去除率36.4%),Cu负载可进一步促进NB降解(降解率77.9%)和TOC的去除(去除率62.3%),相比于单独臭氧氧化体系(NB降解率33.7%,TOC去除率15.7%),处理效果均有明显提升〔95-96〕;在NB及其中间产物降解过程中,·OH发挥重要作用.LeiZhao等〔97〕将金属(Zn,Ni,Fe)负载至蜂窝陶瓷明显提高蜂窝陶瓷催化臭氧氧化NB的活性,且随着负载量(0~8%)的提高,催化硝基苯降解效率提高,其中Fe-CH有最高的催化活性.对其机理进行研究,发现金属的掺杂提高蜂窝陶瓷的pHpzc,从而提高表面羟基-OH2+的密度,而表面-OH2+正是O3吸附和分解产生·OH的活性位点.图 15显示了该研究可能的反应机理. ...
Enhancement mechanism of heterogeneous catalytic ozonation by cordierite-supported copper for the degradation of nitrobenzene in aqueous solution
1
2009
... 蜂窝陶瓷(Ceramic Honeycomb,CH)是结构像蜂窝的多孔陶瓷,可由堇青石、莫来石、钛酸铝、氧化锆、碳化硅质、堇青-莫来石等多种材料组成,其中最广泛应用的是具有高熔点、高机械强度的堇青石(2 MgO·2Al2O3·5SiO2).堇青石可促进硝基苯(NB)在臭氧体系中的降解(降解率50%)及体系TOC的去除(去除率36.4%),Cu负载可进一步促进NB降解(降解率77.9%)和TOC的去除(去除率62.3%),相比于单独臭氧氧化体系(NB降解率33.7%,TOC去除率15.7%),处理效果均有明显提升〔95-96〕;在NB及其中间产物降解过程中,·OH发挥重要作用.LeiZhao等〔97〕将金属(Zn,Ni,Fe)负载至蜂窝陶瓷明显提高蜂窝陶瓷催化臭氧氧化NB的活性,且随着负载量(0~8%)的提高,催化硝基苯降解效率提高,其中Fe-CH有最高的催化活性.对其机理进行研究,发现金属的掺杂提高蜂窝陶瓷的pHpzc,从而提高表面羟基-OH2+的密度,而表面-OH2+正是O3吸附和分解产生·OH的活性位点.图 15显示了该研究可能的反应机理. ...
Novel relationship between hydroxyl radical initiation and surface group of ceramic honeycomb supported metals for the catalytic ozonation of nitrobenzene in aqueous solution
1
2009
... 蜂窝陶瓷(Ceramic Honeycomb,CH)是结构像蜂窝的多孔陶瓷,可由堇青石、莫来石、钛酸铝、氧化锆、碳化硅质、堇青-莫来石等多种材料组成,其中最广泛应用的是具有高熔点、高机械强度的堇青石(2 MgO·2Al2O3·5SiO2).堇青石可促进硝基苯(NB)在臭氧体系中的降解(降解率50%)及体系TOC的去除(去除率36.4%),Cu负载可进一步促进NB降解(降解率77.9%)和TOC的去除(去除率62.3%),相比于单独臭氧氧化体系(NB降解率33.7%,TOC去除率15.7%),处理效果均有明显提升〔95-96〕;在NB及其中间产物降解过程中,·OH发挥重要作用.LeiZhao等〔97〕将金属(Zn,Ni,Fe)负载至蜂窝陶瓷明显提高蜂窝陶瓷催化臭氧氧化NB的活性,且随着负载量(0~8%)的提高,催化硝基苯降解效率提高,其中Fe-CH有最高的催化活性.对其机理进行研究,发现金属的掺杂提高蜂窝陶瓷的pHpzc,从而提高表面羟基-OH2+的密度,而表面-OH2+正是O3吸附和分解产生·OH的活性位点.图 15显示了该研究可能的反应机理. ...
Mechanism of influence of initial pH on the degradation of nitrobenzene in aqueous solution by ceramic honeycomb catalytic ozonation
1
2008
... 在另一项工作中,Lei Zhao等〔98〕研究反应初始pH对反应体系的影响,发现反应溶液初始pH影响溶液中臭氧浓度及臭氧的利用率;pH在pHpzc附近时,蜂窝陶瓷催化臭氧氧化NB的效率最高,此时表面羟基是电中性的,并且表面羟基密度也具有最高值.除此之外,Lei Zhao等〔81, 99〕在另外的两项工作中还分别研究负载Mn及Cu和Mn的蜂窝陶瓷在催化臭氧氧化NB中的应用,研究发现Mn和Cu的掺杂同样提高催化剂表面的羟基浓度,从而促进·OH的产生,致使蜂窝陶瓷的催化活性有所提高. ...
Catalytic ozonation for the degradation of nitrobenzene in aqueous solution by ceramic honeycombsupported manganese
1
2008
... 在另一项工作中,Lei Zhao等〔98〕研究反应初始pH对反应体系的影响,发现反应溶液初始pH影响溶液中臭氧浓度及臭氧的利用率;pH在pHpzc附近时,蜂窝陶瓷催化臭氧氧化NB的效率最高,此时表面羟基是电中性的,并且表面羟基密度也具有最高值.除此之外,Lei Zhao等〔81, 99〕在另外的两项工作中还分别研究负载Mn及Cu和Mn的蜂窝陶瓷在催化臭氧氧化NB中的应用,研究发现Mn和Cu的掺杂同样提高催化剂表面的羟基浓度,从而促进·OH的产生,致使蜂窝陶瓷的催化活性有所提高. ...
Mechanism of enhanced diclofenac mineralization by catalytic ozonation over iron silicate-loaded pumice
2
2017
... 浮石(PMC)是一种火山活动中产生的多孔天然材料,它的主要成分是硅、铝、铁的氧化物.Lei Yuan等〔82〕使用天然浮石作为催化臭氧氧化对氯硝基苯(pCNB)的催化剂,通过研究发现,其主要成分是SiO2、Al2O3、Fe2O3、K2O、MgO、Na2O、TiO2,在pCNB降解过程中,·OH是主要的ROS,不带电的表面羟基基团更有利于·OH的产生.在另一项研究中,Guoying Gao等〔100〕使用负载硅酸铁的浮石(FSO/PMC)催化臭氧氧化双氯芬酸(DCF),加入的FSO/PMC促进了O3的传质和溶解,从而促进·OH的产生;除此之外,FSO/PMC促进H2O2的产生与消耗,同样有利于·OH的产生. ...
... Natural mineral oxide and their catalytic performance
Table 5 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Z1000H〔n(SiO2)/n(Al2O3)=100〕 | 布洛芬 | 布洛芬:60%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | 未提及 | 〔83〕 |
| Z25H〔n(SiO2)/n(Al2O3)=25 | | 布洛芬:78%(CO,30 min),40%(SO,30 min) | O3 1 mg/(min·L);布洛芬15 mg/L;催化剂10 g/L;pH 7.2 | | |
| Ce0.1Fe0.9OOH | 磺胺二甲嘧啶(SMT) | TOC:42.1%(CO,120 min),24.2%(SO,120 min) | O3 15 L/min;SMT 20 mg/L;催化剂0.2 g/L;pH 7.3 | 表面羟基是O3吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔89-90〕 |
| Ce0.1Fe0.9OOH | 邻苯二甲酸二甲酯(DMP) | DMP:100%(CO,30 min),80%(SO,30 min)TOC:40%(CO,60 min),15%(SO,60 min) | O3 15 mg/L,0.4 L/min;DMP 10 mmol/L;催化剂0.2 g/L;pH 6.8 | 表面羟基是O3的吸附位点,Fe2+/Fe3+和Ce3+/Ce4+氧化还原过程促进O3分解 | 〔88〕 |
| α-Fe0.9Mn0.1OOH | 碘海醇 | 碘海醇:87.6%(CO,15 min),70%(SO,15 min) | O3 20.8μmol/L;碘海醇1.22μmol/L;催化剂0.075 g/L;pH 7.0 | 表面Lewis酸性位点;表面羟基及表面氧空位 | 〔91-92〕 |
| Co-FeOOH | 阿提洛尔(ATL) | ATL:98%(CO,10 min),85%(SO,10 min);TOC:35%(CO,60 min),21%(SO,60 min) | O3 0.15 mg/L;ATL 200 μg/L;催化剂0.1 g/L;pH 7.0±0.1 | 表面羟基 | 〔93〕 |
| PMC | 对氯硝基苯(pCNB) | pCNB:85%(CO,20 min),55%(SO,20 min)TOC:32%(CO,20 min),15%(SO,20 min) | O3 0.6 mg/L;pCNB 100 μg/L;催化剂1 g/L;pH 6.86 | 不带电的表面羟基(pH≈pHpzc) | 〔82〕 |
| FSO/PMC | DCF | TOC:73.3%(CO,60 min),32.3%(SO,60 min) | O3 5.52 mg/L;DCF 29.6 mg/L;催化剂0.8 g/L;pH 7.0 | 不带电的表面羟基促进O3的传质与溶解 | 〔100〕 |
5 其他多金属氧化物多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Catalytic ozonation of Acid Red B in aqueous solution over a Fe-Cu-O catalyst
2
2013
... 多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
... Other polymetallic oxides and their catalytic performance
Table 6 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Fe-Cu-O | 酸性红B(ARB) | ARB:90%(CO,60 min),66%(SO);COD:70%(CO,60 min),48%(SO) | O3 6 mg/min,100 mL/min;ARB 200 mg/L;催化剂1 g/L;pH 6.36 | 表面羟基 | 〔101〕 |
| Mn-Ce-O〔n(Mn)/n(Ce)=8/2〕 | 安替比林(AP) | AP:100%(CO,2 min),100%(SO,5 min);TOC:87%(CO,60 min),44%(SO,60 min) | O3 20 mg/L,200 mL/min;AP 40 mg/L;催化剂0.2 g/L;pH 6.5 | 表面电子传递 | 〔16〕 |
| Mn-Co-Fe-O | 对硝基苯酚(PNP) | PNP:100%(CO,15 min),100%(SO,60 min);TOC:95%(SO,60 min),50%(SO,60 min) | O3 2 mg/min;PNP 10 mg/L;催化剂0.1 g/L;pH 6.5 | 去质子化表面羟基(pH>pHpzc) | 〔103〕 |
| CaMn4O8 | 对硝基苯酚(PNP) | PNP:100%(CO,30 min),100%(SO,60 min) | O3 100 mg/min,50 mg/L;PNP 50 mg/L;催化剂0.1 g/L;pH 5.7 | Mn3+/Mn4+氧化还原循环促进O3分解 | 〔15〕 |
注: CO是催化臭氧氧化过程,SO是单独臭氧氧化过程. ...
Influence of the surface hydroxyl groups of MnOx/SBA-15 on heterogeneous catalytic ozonation of oxalic acid
1
2014
... 多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
Catalytic ozonation of pnitrophenol over mesoporous Mn-Co-Fe oxide
2
2014
... 多金属混合氧化物有类似尖晶石多金属氧化物的组成,但它们有不同的结构.Fe-Cu-O混合金属氧化物可有效催化臭氧氧化酸性红B,60 min内色度和COD的去除率分别为90%和70%,远远高于臭氧氧化的效率(66%和48%)〔101〕.Mn-Ce-O也可促进O3分解,Shengtao Xing等〔16〕认为Mn-Ce-O促进O3分解产生活性氧物种是安替比林(AP)矿化的原因,通过对比不同n(Mn)/n(Ce)的Mn-Ce-O氧化物的催化活性发现,Mn-Ce-O催化活性取决于它们的电子传递能力,这在很多研究中被证实是促进臭氧产生活性氧物种的机理之一〔21, 102〕.Zichuan Ma等〔103〕通过氧化-沉淀法合成Mn-Co-Fe混合金属氧化物用于催化臭氧氧化对硝基苯酚(PNP),发现催化剂的活性取决于表面性质,带负电的催化剂表面促进O3的吸附和分解,而带正电的催化剂表面有利于有机中间体的吸附.Yuxian Wang等〔15〕以碳酸钙和碳酸锰为前驱体,采用简单的共沉淀方法,合成CaMn3O6和CaMn4O8混合价锰酸钙并用于催化臭氧氧化4-硝基苯酚,相比于MnO2,CaMn3O6和CaMn4O8催化臭氧氧化4-硝基苯酚降解的速率及TOC去除率更高,且Ca显著稳定Ca-Mn-O结构,使Mn离子在反应液中的溶出量小于2 mg/L,远低于MnO2的Mn离子溶出量(10 mg/L).该研究认为单线态氧1O2、O2·-及O3是主要的活性物种,且起主导作用的含氧活性物种是O2·-. ...
... Other polymetallic oxides and their catalytic performance
Table 6 | 催化剂 | 污染物 | 处理效率 | 反应条件 | 活性位点 | 文献 |
| Fe-Cu-O | 酸性红B(ARB) | ARB:90%(CO,60 min),66%(SO);COD:70%(CO,60 min),48%(SO) | O3 6 mg/min,100 mL/min;ARB 200 mg/L;催化剂1 g/L;pH 6.36 | 表面羟基 | 〔101〕 |
| Mn-Ce-O〔n(Mn)/n(Ce)=8/2〕 | 安替比林(AP) | AP:100%(CO,2 min),100%(SO,5 min);TOC:87%(CO,60 min),44%(SO,60 min) | O3 20 mg/L,200 mL/min;AP 40 mg/L;催化剂0.2 g/L;pH 6.5 | 表面电子传递 | 〔16〕 |
| Mn-Co-Fe-O | 对硝基苯酚(PNP) | PNP:100%(CO,15 min),100%(SO,60 min);TOC:95%(SO,60 min),50%(SO,60 min) | O3 2 mg/min;PNP 10 mg/L;催化剂0.1 g/L;pH 6.5 | 去质子化表面羟基(pH>pHpzc) | 〔103〕 |
| CaMn4O8 | 对硝基苯酚(PNP) | PNP:100%(CO,30 min),100%(SO,60 min) | O3 100 mg/min,50 mg/L;PNP 50 mg/L;催化剂0.1 g/L;pH 5.7 | Mn3+/Mn4+氧化还原循环促进O3分解 | 〔15〕 |
注: CO是催化臭氧氧化过程,SO是单独臭氧氧化过程. ...



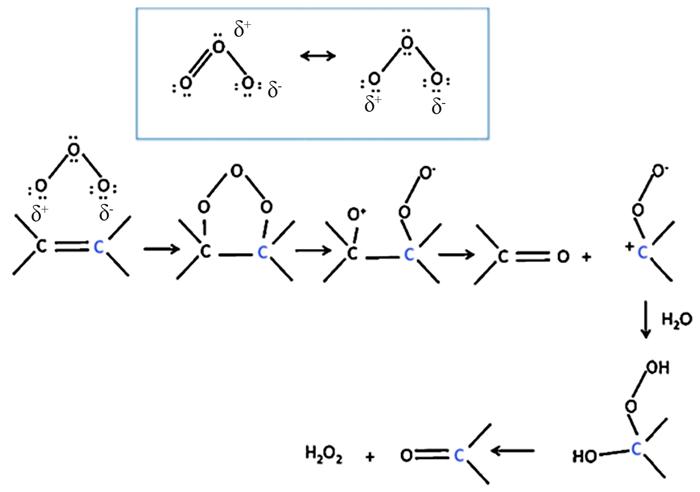

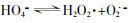

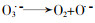
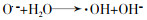


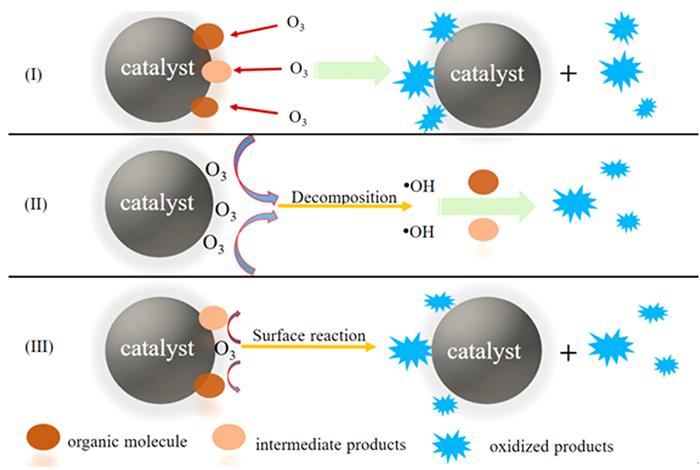
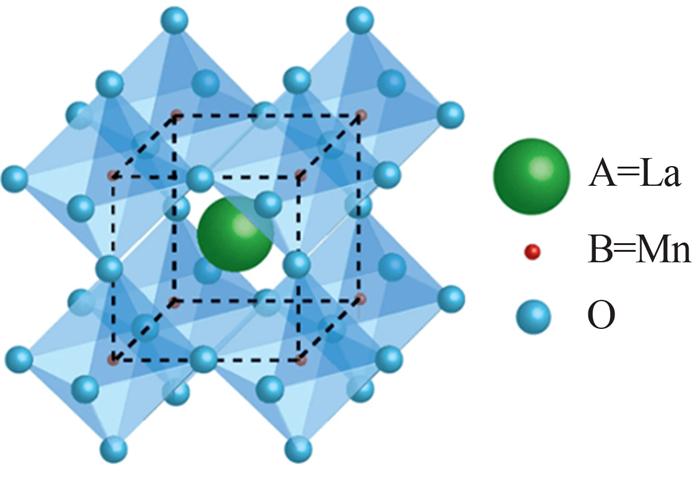
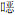
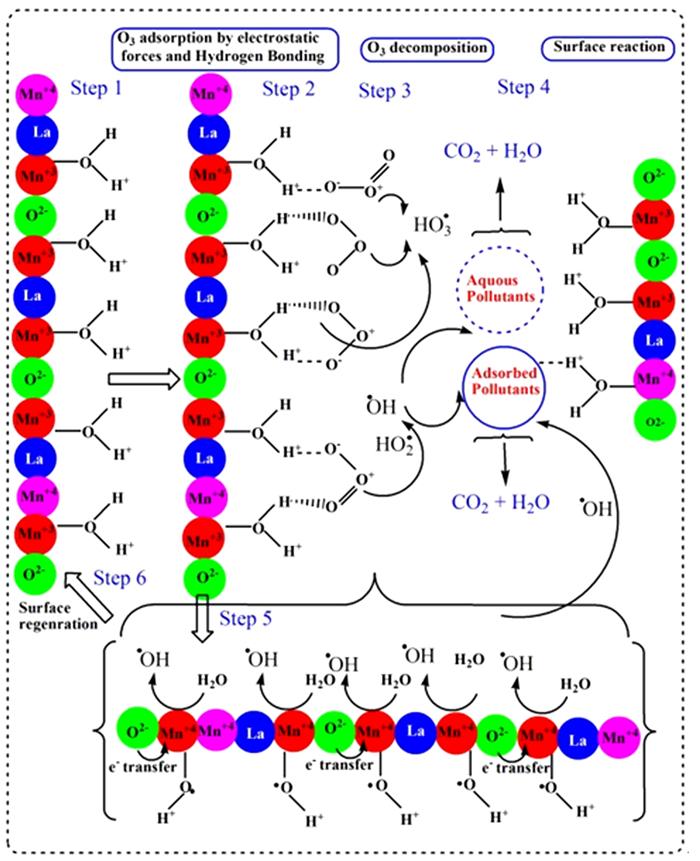
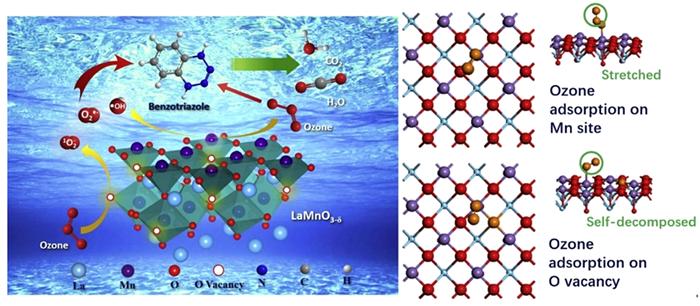
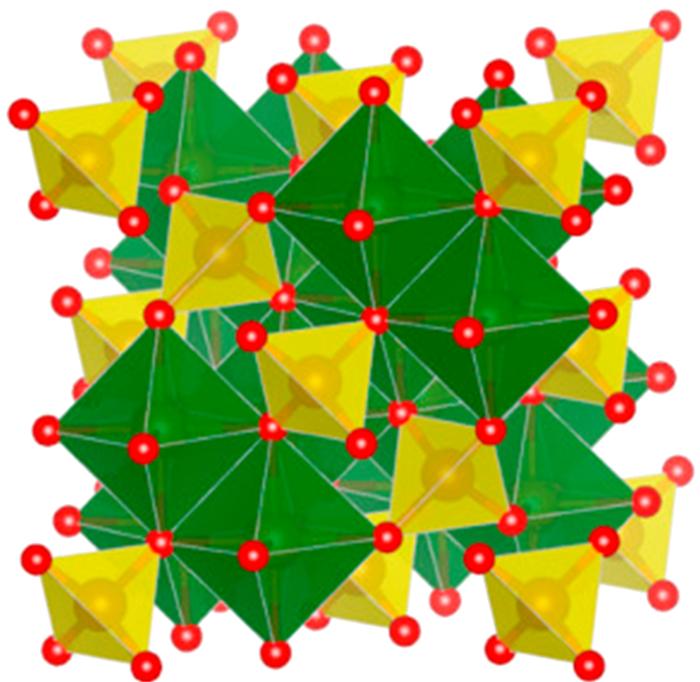
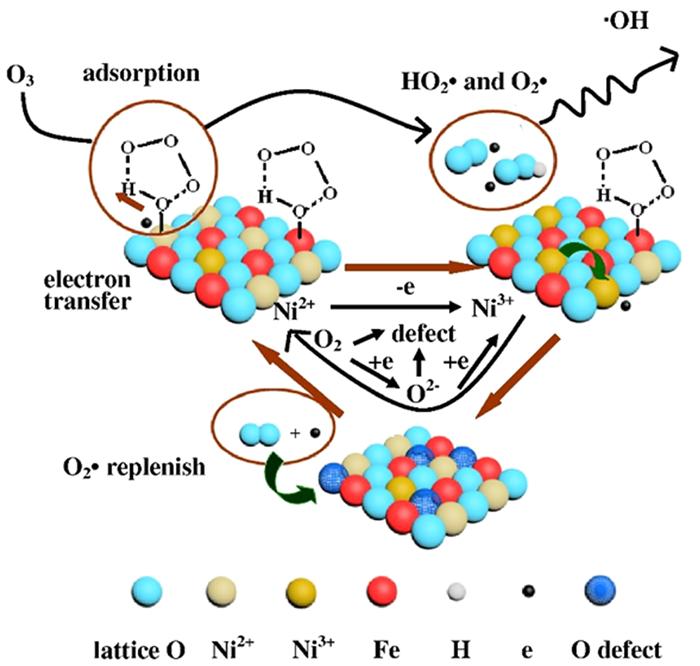
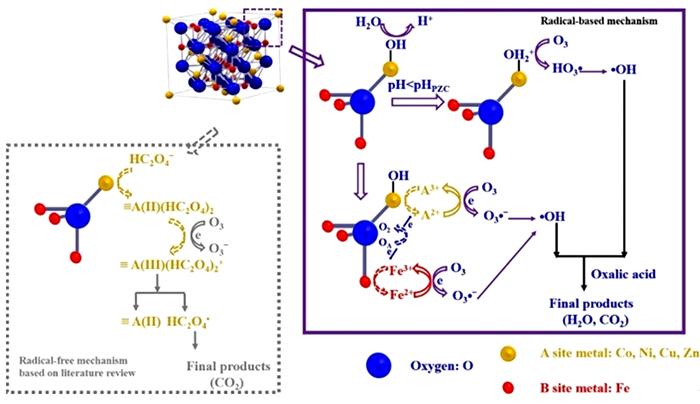
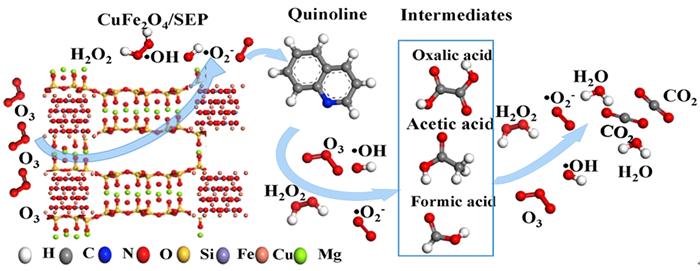
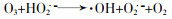
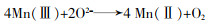
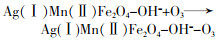
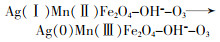
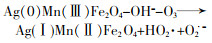
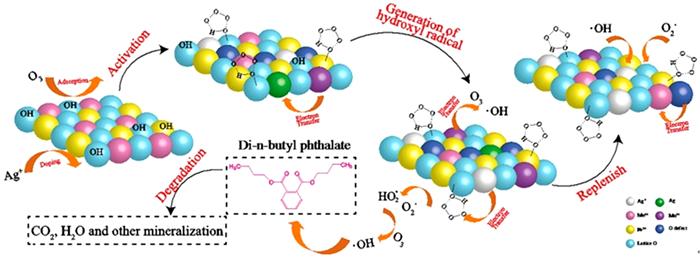
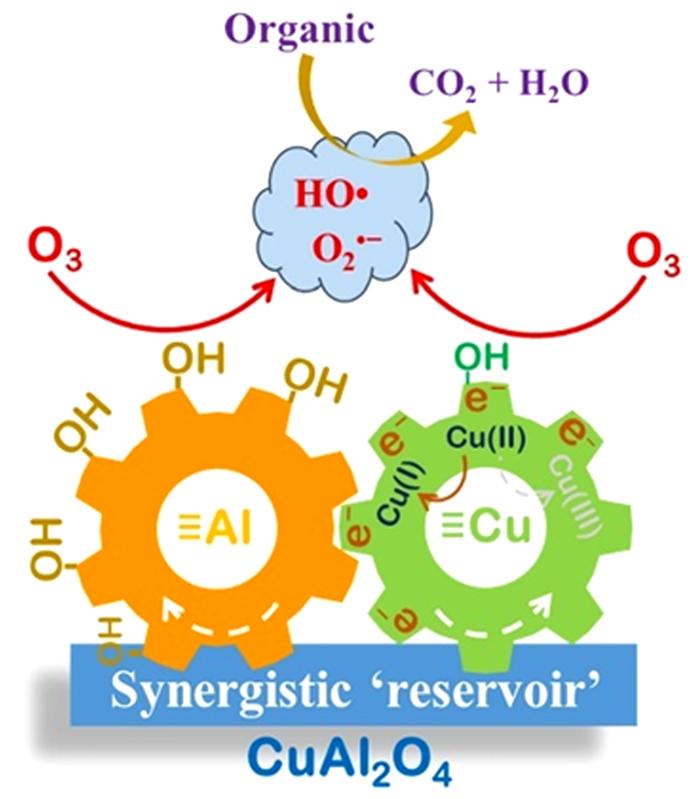
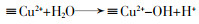
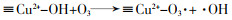

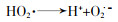

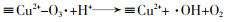

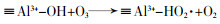
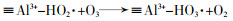
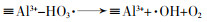
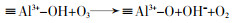

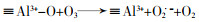
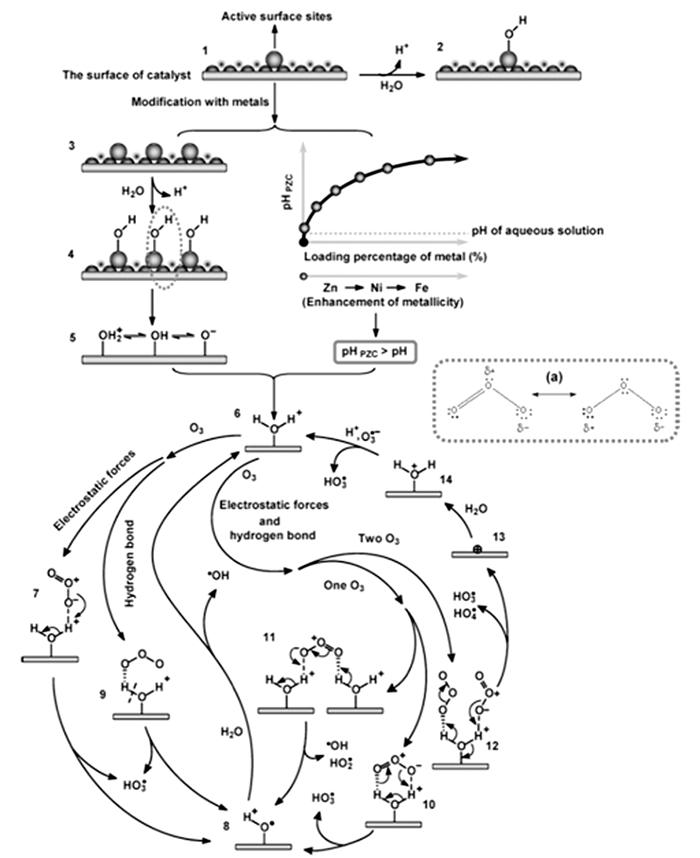


 津公网安备 12010602120337号
津公网安备 12010602120337号{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}